
آنالیز توالی ژنتیکی بیماری های خون ریزی دهنده ارثی
ژن های کدکننده ی پروتئین های فاکتور انعقادی (کوآگولاسیون) در بین اولین ژن های انسانی قرار دارند که در طی 25 سال گذشته تعیین ویژگی شده اند. از آن زمان تا حالا، پیشرفت قابل توجهی در کاربردی سازی این اطلاعات برای 2 تا از معمول ترین اختلالات شدید خون ریزی دهنده ی ارثی، یعنی هموفیلی A و B صورت گرفته است. برای این اختلالات وابسته به X، تعیین خصوصیت ژنتیکی جهش های ایجاد کننده ی بیماری، حالا به درون استانداردهای مراقبتی اضافه شده است و اطلاعات ژنتیکی برای طبقه بندی ریسک عوارض درمانی مورد استفاده قرار گرفته اند. از روی داده پایگاه های الکترونیکی که بیشتر از 2100 جهش منحصر به فرد برای هموفیلی A و بیشتر از 1100 جهش برای هموفیلی B را به تفصیل بیان کرده اند، می توان نتیجه گرفت که این بیماری ها در بین گسترده ترین اختلالات ارثی در انسان ها قرار دارند که تعیین ویژگی شده اند. آزمایش ژنتیکی بیماری های خون ریزی دهنده ی نادر، همانطور که انتظارش می رود، پیشرفت کمی داشته است. به هرحال، اینجا بازهم، پایگاه داده های الکترونیکی جهش ایجاد شده اند و رهنمودهای عالی را برای کاربرد آنالیزهای ژنتیکی، به عنوان یک روش تاییدی برای تشخیص، ارائه کرده اند. اخیرا، پیشرفت هایی در زمینه ی شناسایی لوکوس های جهش یافته در یک طیفی از اختلالات ارثی پلاکت خون انجام شده است و این یافته ها آغاز به کاربرد برای تشخیص ژنتیکی این شرایط کرده اند. بررسی بیمارانی که فنوتیپ خون ریزی دهنده داشتند اما با استفاده از استراتژی های ژنوم گسترده تشخیص داده نشده بودند، ممکن است ژن های جدیدی را شناسایی کند که قبلا مشخص نشده بود که یک نقشی را در هموستازی دارند (خون. 2013، 122(20):3423-3431).
استراتژی های ژنومی آینده برای تعیین خصوصیت اختلالات خون ریزی دهنده ی ارثی
همانطور که تکنولوژی های ژنومی به پیشرفت خود ادامه می دهند، پتانسیل این استراتژی ها برای الحاق به درون الگوریتم های تشخیصی نیز قطعا افزایش پیدا می کند. به هرحال، قبل از اینکه این امر بتواند انجام شود، یک تعدادی از ضمیمه های مهم به فراساختار تشخیصی اخیر ما باید اضافه شود.
پایگاه داده هایی که به دقت برگزیده شده اند، مانند Reactome (www.reactome.org)، اطلاعات مهمی را فراهم می کنند که پزشکان و دانشمندان را قادر می سازد تا به اطلاعات درستی در مورد مسیرهای سلولی و تجسم روابط بین واریانت های توالی نادر و فنوتیپ های بالینی دست پیدا کنند. پایگاه داده ی GEN2PHEN (www.gen2phen.org) ازطریق یک همکاری بین مرکز ملی برای بیوانفورماتیک و موسسه ی بیوانفورماتیک اروپا ایجاد شده است و سایر تلاش ها منجر به ایجاد سایت لوکوس ژنوم مرجع (www.Irg-sequence.org) شده اند، این سیستم، یک توالی DNAیی ژنومی را ارائه می کند که نماینده ی یک ژن منفرد است که محتوای مرکزی آن هرگز تغییر نمی کند و یک لایه ی حاشیه ای قابل به روز رسانی دارد که شامل واریانت های توالی است.
The genes encoding the coagulation factor proteins were among the first human genes to be characterized over 25 years ago. Since then, significant progress has been made in the translational application of this information for the 2 commonest severe inherited bleeding disorders, hemophilia A and B. For these X-linked disorders, genetic characterization of the disease-causing mutations is now incorporated into the standard of care and genetic information is used for risk stratification of treatment complications. With electronic databases detailing >2100 unique mutations for hemophilia A and >1100 mutations for hemophilia B, these diseases are among the most extensively characterized inherited diseases in humans. Experience with the genetics of the rare bleeding disorders is, as expected, less well advanced. However, here again, electronic mutation databases have been developed and provide excellent guidance for the application of genetic analysis as a confirmatory approach to diagnosis. Most recently, progress has also been made in identifying the mutant loci in a variety of inherited platelet disorders, and these findings are beginning to be applied to the genetic diagnosis of these conditions. Investigation of patients with bleeding phenotypes without a diagnosis, using genome-wide strategies, may identify novel genes not previously recognized as playing a role in hemostasis. (Blood. 2013; 122(20):3423-3431)
Future genomic strategies for the characterization of inherited bleeding disorders
As genomic technologies continue to advance, the potential for incorporating these strategies into diagnostic algorithms will inevitably increase. However, before this can happen, a number of critical additions to our current diagnostic infrastructure must be developed.
Carefully curated databases, such as Reactome (www.reactome. org), provide key information that will enable physicians and scientists to access relevant information on cellular pathways and to visualize the relationships between rare sequence variants and clinical phenotypes. The GEN2PHEN database (www.gen2phen.org), created through a collaboration between the National Center for Bioinformatics and European Bioinformatics Institute, and other efforts have resulted in the establishment of the Locus Reference Genome (www.lrg-sequence. org), a system that provides a genomic DNA sequence representing a single gene, whose core content never changes, with an updateable annotation layer that includes sequence variants.


(جهت بزرگ نمایی روی عکس کلیک نمایید)
هموفیلی A و B
هموفیلی B leiden
ژنوتیپ های متفاوت – تست فاکتور VIII
VWD
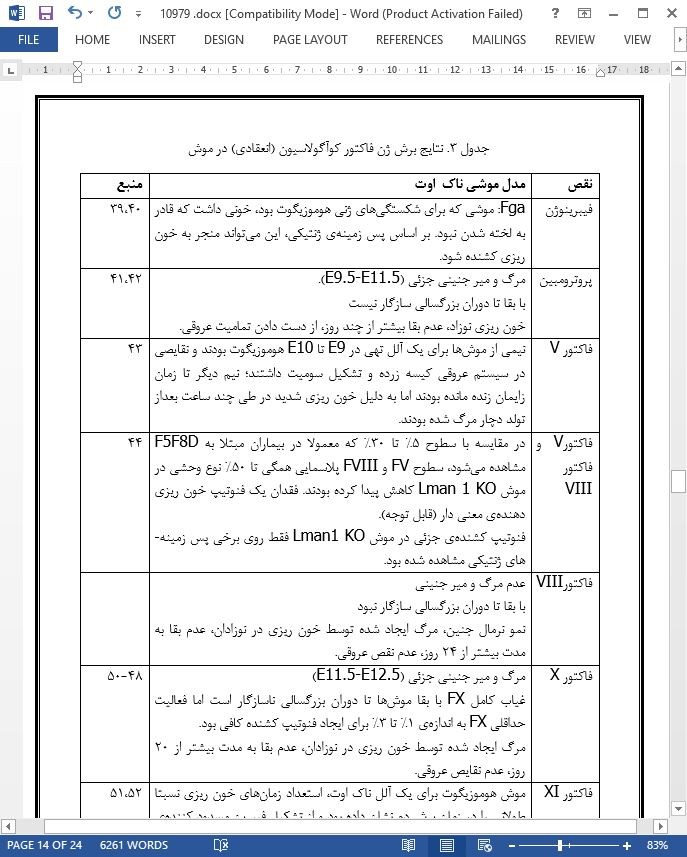
نقایص نادر فاکتور کوآگولاسیون (انعقاد)
طیف جهشی اختلالات خون ریزی دهنده ی نادر
چالش های باقی مانده در ویژگی یابی ژنتیکی نقایص نادر فاکتور کوآگولاسیون (انعقادی)
اختلالات خون ریزی دهنده ی ارثی مرتبط با پلاکت ها
1. واریانت های معمول با اثرات کوچک
2. واریانت های نادر با اثرات بزرگ
تعیین خصوصیات اختلالات ارثی پلاکت
اختلال پلاکت خانوادگی همراه با لوسمی میلوئیدی حاد
سندرم هرمانسکی – پودلاک (HPS)
ترومبوسیتوپنی 2، که تحت عنوان ترومبوسیتوپنی مرتبط با دمین تکرار آنکرین 26 (ANKRD26) نیز شناخته می شود
سندرم ترومبوسیتوپنی همراه با فقدان رادیوس
سندرم آلدریچ ویسکوت (WAS)
استراتژی های ژنومی آینده برای تعیین خصوصیت اختلالات خون ریزی دهنده ی ارثی
Hemophilia A and B
Hemophilia B Leiden
Factor VIII assay–discrepant genotypes
VWD
Rare coagulation factor deficiencies
Mutational spectrum of rare bleeding disorders
Remaining challenges in the genetic characterization of rare coagulation factor deficiencies
Inherited platelet-related bleeding disorders
1. Common variants with small effects
2. Rare variants with large effects
Genetic characterization of inherited platelet disorders
Familial platelet disorder with predisposition to acute myeloid leukemia
Hermansky-Pudlak syndrome (HPS)
Gray platelet syndrome
Thrombocytopenia 2, also known as ankyrin repeat domain 26- (ANKRD26)-related thrombocytopenia (ANKRD26-RT)
Thrombocytopenia and absent radii syndrome
Wiskott-Aldrich syndrome (WAS)
Future genomic strategies for the characterization of inherited bleeding disorders
- اصل مقاله انگلیسی با فرمت ورد (word) با قابلیت ویرایش
- ترجمه فارسی مقاله با فرمت ورد (word) با قابلیت ویرایش، بدون آرم سایت ای ترجمه
- ترجمه فارسی مقاله با فرمت pdf، بدون آرم سایت ای ترجمه