
فعال سازی میکروگلیا و مسیر نیتریک اکسید PKG/cGMP/در مالتیپل اسکلروزیس تجربی
چکیده
نشان داده شده است که طی بیماری ام اس، ارتباط نزدیکی میان التهاب و دژنراسیون عصبی آکسونی بهوجود میآید که به فرضیهای منتهی میشود مبنی بر اینکه سازوکارهای ایمنی میتوانند باعث تحلیل عصبی و پیشرفت غیر قابل بازگشت بیماری شوند. بهنظر میرسد کمبود انرژی و التهاب در اثر اختلال عملکرد میتوکندری در این فرایند نقش دارند. ما در این مطالعه با ثبت الکتروفیزیولوژیک پتانسیل میدانی جسم مخطط ، چگونگی تاثیر فرایند التهابی مربوط به آنسفالومیلیت خود ایمن تجربی (EAE) بر آسیبپذیری عصبی نسبت به مهار کمپلکس IV میتوکندری (که یکی از اجزای حیاتی برای فعالیت میتوکندری و مسئول استفاده از حدود %90 اکسیژن سلولی است) را بررسی کردیم. ما نشان دادیم که طی مرحله عود EAE حاد، حساسیت نورونی نسبت به مهار کمپلکس IV بهطور چشمگیری افزایش پیدا میکند. این اثر مخرب، با مهار دارویی میکروگلیا، سنتز نیتریک اکسید (NO) و مسیر داخل سلولی آن (شامل گوانیلیل سیکلاز محلول sGC)) و پروتئینکیناز G (PKG)) خنثی میشود. نتایج بهدست آمده نشان میدهد کمپلکس IV میتوکندری نقش مهمی در حفظ هومئوستازی انرژتیک نورونی طی آنسفالومیلیت خود ایمن تجربی ایفا میکند. فرایندهای آسیبشناختی مربوط به ام اس تجربی و بهخصوص فعال شدن میکروگلیا و مسیر نیتریک اکسید، باعث افزایش آسیبپذیری نورونی نسبت به مهار کمپلکس IV میتوکندری میشود که اهداف دارویی امیدبخشی را نشان میدهد.
1. مقدمه
اسکلروز چندگانه (MS) یکی از اختلالات مزمن سیستم عصبی مرکزی (CNS) است که در سراسر جهان پراکندگی داشته و بزرگسالان را در سنین جوانی تحت تأثیر قرار میدهد و بهطور بالقوه باعث تجمع پیشرونده ناتوانی عصبی شده و بر کیفیت و امید به زندگی بیماران تأثیر میگذارد. از همان مراحل نخست بیماری اماس، فرآیندهای التهابی، هم بهصورت حملات ایمنی مجدد از طریق سد خونی مغزی از محیط به سیستم عصبی مرکزی و هم فعالسازی واکنشهای ایمنی ذاتی مستقیم در خود سیستم عصبی مرکزی مانند سلولهای میکروگلیا، در تشخیص آسیبزایی آن مؤثر هستند. از همان مراحل نخست بیماری، فرایند التهابی همراه با تحلیل رفتن آکسون نورونها در مغز بیماران مبتلا به اماس قابل شناسایی بوده و نشاندهنده یکی از دلایل اصلی افزایش پیشرونده ناتوانی عصبی است. در این سناریو، شناسایی ارتباط آسیبزایی میان فرآیندهای التهابی و تحلیلبرنده عصبی طی بیماری ام اس میتواند برای توسعه درمانهای مؤثر علیه افزایش ناتوانی مفید باشد. مطالعات سالهای اخیر نشان دادهاند که اختلال عملکرد میتوکندری در میانجیگری تحلیل عصبی آکسونی و پیشرفت غیر قابل برگشت بیماری طی این دوره، نقش آسیبزای بالقوهای دارد. میتوکندریها اندامکهای داخل سلولی هستند که بخش عمده آدنوزینتریفسفات (ATP) سلول را تولید میکنند، اما علاوهبراین، در متابولیسم کلسیم، تولید گونههای واکنشدهنده اکسیژن (ROS) و آپوپتوز سلولی نقش دارند.
5. نتایج
به نظر میرسد التهاب و اختلال عملکرد میتوکندری در یک چرخه معیوب با یکدیگر ارتباط دارند که درنهایت با روند همافزایی، همسو میشوند که مسئول دژنراسیون عصبی آکسونی در طی دوره اماس است. دادههای بهدست آمده این شواهد را تأیید میکنند که میکروگلیا همراه با فعالسازی مسیر NO در پیوند دادن روند التهاب مرتبط با اماس تجربی، با آسیبپذیری عصبی آزمایششده مرتبط با اختلال عملکرد میتوکندری میتواند نقش کلیدی ایفا نماید.
سنتز NO مرتبط با التهاب و فعالیت حفظشده مسیر داخل سلولی آن شامل sGC و PKG میتواند نشاندهنده یک هدف دارویی امیدبخش باشد که اثر سینرژیک و مخرب التهاب و اختلال عملکرد میتوکندری را خنثی کرده و راهبردهای مؤثری را بهمنظور محافظت عصبی تعیین نماید.
Abstract
During multiple sclerosis (MS), a close link has been demonstrated to occur between inflammation and neuro-axonal degeneration, leading to the hypothesis that immune mechanisms may promote neurodegeneration, leading to irreversible disease progression. Energy deficits and inflammation-driven mitochondrial dysfunction seem to be involved in this process. In this work we investigated, by the use of striatal electrophysiological field-potential recordings, if the inflammatory process associated with experimental autoimmune encephalomyelitis (EAE) is able to influence neuronal vulnerability to the blockade of mitochondrial complex IV, a crucial component for mitochondrial activity responsible of about 90% of total cellular oxygen consumption. We showed that during the acute relapsing phase of EAE, neuronal susceptibility to mitochondrial complex IV inhibition is markedly enhanced. This detrimental effect was counteracted by the pharmacological inhibition of microglia, of nitric oxide (NO) synthesis and its intracellular pathway (involving soluble guanylyl cyclase, sGC, and protein kinase G, PKG). The obtained results suggest that mitochondrial complex IV exerts an important role in maintaining neuronal energetic homeostasis during EAE. The pathological processes associated with experimental MS, and in particular the activation of microglia and of the NO pathway, lead to an increased neuronal vulnerability to mitochondrial complex IV inhibition, representing promising pharmacological targets.
1. Introduction
Multiple Sclerosis (MS) is a worldwide-diffused chronic central nervous system (CNS) disorder, affecting young adults and potentially leading to progressive accumulation of neurological disability, with a relevant impact on patient's quality of life and life expectancy (Ontaneda et al., 2017). Inflammatory processes characterize MS pathogenesis since its earliest phases, both in the form of recurrent immune attacks from the periphery to the CNS through the blood-brain barrier and via the activation of direct innate immune responses within the CNS itself, e.g. microglial cells. Together with inflammatory processes, neuro-axonal degeneration can be detected in the CNS of patients affected by MS since the earliest phases of the disease (Dutta and Trapp, 2007; Trapp and Nave, 2008) and it represents one of the major causes underlying the progressive accumulation of neurological disability (Hauser and Oksenberg, 2006; Trapp and Nave, 2008). In this scenario, the identification of the pathogenic link between inflammatory and neurodegenerative processes during MS (Geurts et al., 2009) could help in the development of effective therapies against disability accumulation. During last years, several studies suggested a potential pathogenic role for mitochondrial dysfunction in mediating neuro-axonal degeneration and irreversible disease progression during the course of MS (Calabrese et al., 2015; Campbell et al., 2014; Di Filippo et al., 2010; Witte et al., 2014). Mitochondria are intracellular organelles responsible for the majority of adenosine triphosphate (ATP) cellular production, but they also play a role in calcium metabolism, reactive oxygen species (ROS) production and cellular apoptosis (Andreyev et al., 2005; Di Mauro and Schon, 2003; Taylor and Turnbull, 2005).
5. Conclusions
Inflammation and mitochondrial dysfunction seem to be intertwined in a vicious cycle, ultimately converging into a common synergistic process responsible for neuro-axonal degeneration during the course of MS. The obtained data support the evidence that microglia could play a key role, together with the activation of the NO pathway, in linking the inflammatory process associated with experimental MS to the observed neuronal susceptibility to mitochondrial dysfunction.
Inflammation-related NO synthesis and the sustained activation of its intracellular pathway, involving sGC and PKG may represent a promising pharmacological target to counteract the synergic detrimental effect of inflammation and mitochondrial dysfunction (Colombo et al., 2012, 2014) and to design effective neuro-protective strategies.


(جهت بزرگ نمایی روی عکس کلیک نمایید)
چکیده
1. مقدمه
2. مواد و روشها
2.1. القای آنسفالومیلیت خودایمن تجربی مزمن عودکننده
2.2. آمادهسازی و نگهداری قطعات نمونه برای سنجش الکتروفیزیولوژی
2.3. الکتروفیزیولوژی
2.4. ایمونوهیستوشیمی
2.5. آمادهسازی برشهای خام میتوکندری
2.6. ارزیابی فعالیت کمپلکس IV
2.7. داروها
2.8. تجزیه و تحلیل آماری
3. نتایج
3.1. سمیت عصبی القاشده توسط مهار کمپلکس IV زنجیره تنفسی میتوکندری، بهطور قابلتوجهی در طی ام اس تجربی افزایش پیدا میکند
3.2. سدیم آزید NaN3 موجب مهار قابلتوجه کمپلکس IV میتوکندری در شرایط کنترل و در طی EAE میشود.
3.3. افزایش آسیب عصبی مرتبط با مهار کمپلکس IV میتوکندری در طی EAE بستگی به مسیر /cGMP/ PKG نیتریک اکسید دارد
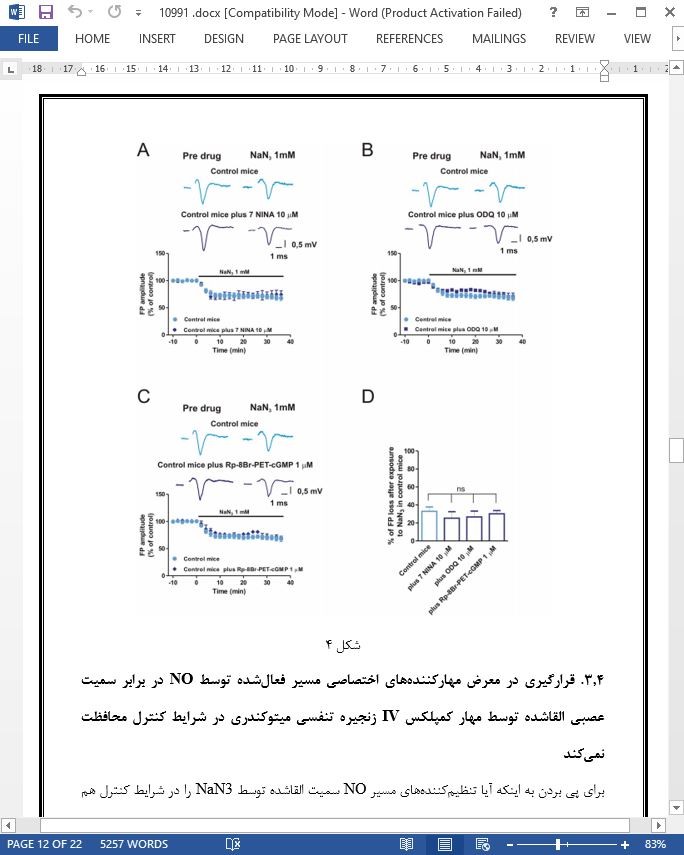
3.4. قرارگیری در معرض مهارکنندههای اختصاصی مسیر فعالشده توسط NO در برابر سمیت عصبی القاشده توسط مهار کمپلکس IV زنجیره تنفسی میتوکندری در شرایط کنترل محافظت نمیکند
3.5. فعالکنندههای اختصاصی مسیر فعال NO سمیت عصبی القاشده توسط مهار کمپلکس IV زنجیره تنفسی میتوکندری در شرایط کنترل را افزایش نمیدهند
3.6. قرارگیری در معرض سایتوکاینهای پیشالتهابی، سمیت عصبی القاشده توسط مهار کمپلکس IV میتوکندری را تحت تأثیر قرار نمیدهد
3.7. فعالسازی میکروگلیا موجب پیشبرد افزایش آسیبپذیری عصبی نسبت به مهار کمپلکس IV میتوکندری طی EAE میشود
4. بحث
5. نتایج
ABSTRACT
1. Introduction
2. Materials and methods
2.1. Induction of chronic-relapsing EAE
2.2. Preparation and maintenance of slices for electrophysiological recordings
2.3. Electrophysiology
2.4. Immunohistochemistry
2.5. Mitochondrial crude fraction preparation
2.6. Complex IV activity evaluation
2.7. Drugs
2.8. Statistical analysis
3. Results
3.1. Neuronal toxicity induced by mitochondrial respiratory chain complex IV inhibition is markedly enhanced during experimental MS
3.2. Sodium azide (NaN3) induces a significant mitochondrial complex IV inhibition both in control conditions and during EAE
3.3. The increased neuronal vulnerability to mitochondrial complex IV inhibition during EAE depends on the nitric oxide/cGMP/PKG pathway
3.4. Exposure to specific inhibitors of NO-activated pathway does not protect from neuronal toxicity induced by mitochondrial respiratory chain complex IV inhibition in control conditions
3.5. Specific activators of the NO-activated pathway do not enhance neuronal toxicity induced by mitochondrial respiratory chain complex IV inhibition in control conditions
3.6. Exposure to pro-inflammatory cytokines does not influence neuronal toxicity induced by mitochondrial complex IV inhibition
3.7. Microglial activation drives the increased neuronal susceptibility to mitochondrial complex IV inhibition during EAE
4. Discussion
5. Conclusions
- ترجمه فارسی مقاله با فرمت ورد (word) با قابلیت ویرایش، بدون آرم سایت ای ترجمه
- ترجمه فارسی مقاله با فرمت pdf، بدون آرم سایت ای ترجمه