
دانلود مقاله ارزیابی تکاملی تمام-ژنومی ویروس کرونای نوظهور فرضیه پدید آمدن آن در نتیجه یک رخداد نوترکیبی اخیر را رد می کند
چکیده
پیش زمینه: اخیراً کروناویروس نوظهوری (2019-nCoV) در ارتباط با انتقال انسان به انسان و عفونت شدید در انسان ها در شهر ووهان چین گزارش شده است. هدف ما توصیف روابط ژنتیکی کروناویروس نوظهور و جست و جو برای یافتن نوترکیبی مفروض در بین زیرسرده ی ساربکوویروس بوده است.
روشها: نوترکیبی مفروض با استفاده از برنامه های RDP4 و Simplot نسخه 3.5.1 مورد بررسی قرار گرفت و خوشه بندی تبارزایشی نامتجانس در قطعات ژنومی واحد با کمک ارزیابی تبارزاشی و با استفاده از بزرگنمایی بیشینه و روش های بیزی تایید گردید.
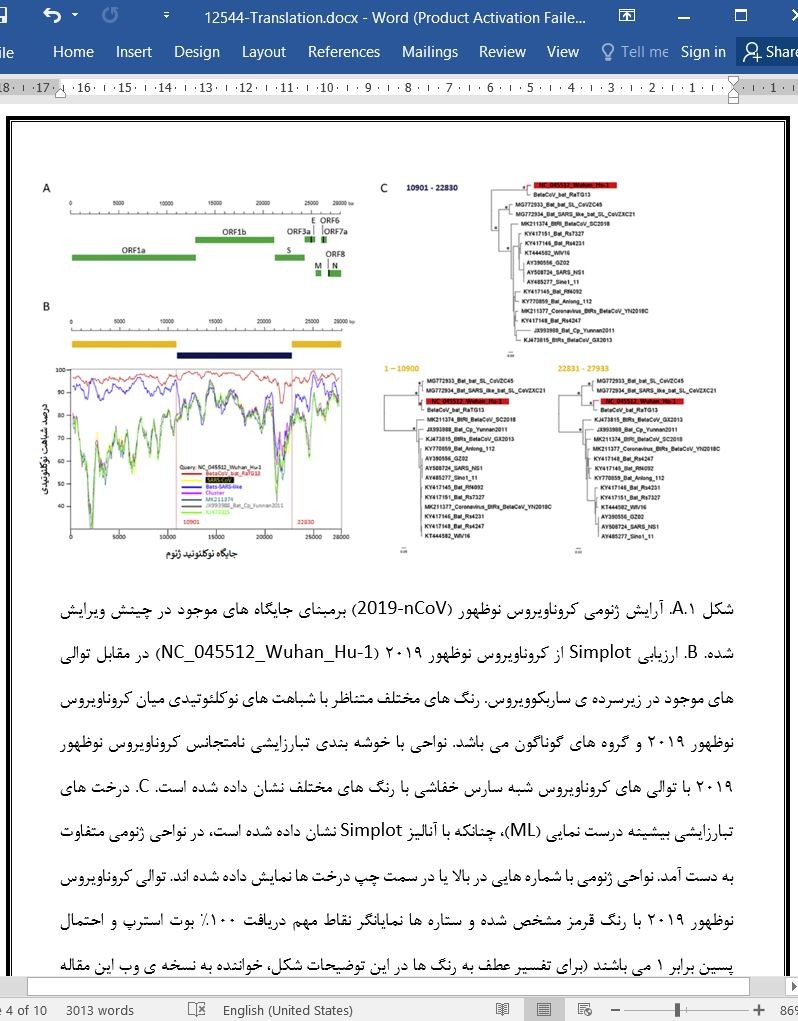
نتایج: ارزیابی ما مطرح کننده آن است که گرچه کروناویروس نوظهور در سراسر ژنوم خود ارتباط نزدیکی با توالی RaTG13 کروناویروس خفاشی دارد (شباهت توالی 96.3%)، خوشه بندی نامتجانسی با توالی های کروناویروس های شبه سارس خفاشی (bat-SARS like) از خود نشان می دهد. به ویژه، در بخش '5 دربردارنده ی 11498 نوکلئوتید اول و بخش نهایی '3 دربردارنده ی جایگاه های 30696-24341، کروناویروس نوظهور و RaTG13 با توالی های کروناویروس شبه سارس خفاشی خوشه ی واحدی تشکیل می دهند، درحالیکه در ناحیه ی میانی شامل انتهای '3 ORF1a، ORF1b و تقریبا نیمی از نواحی زوائد گرزی شکل، کروناویروس نوظهور و RaTG13 در دودمان مجزایی با فاصله از کروناویروس خفاشی در درون شاخه ی ساربکوویروس گروه بندی شده اند.
جمع بندی: سطوح شباهت ژنتیکی در بین کروناویروس نوظهور 2019 و RaTG13 بیانگر آن است که دومی نوعِ بخصوصی که عامل همه گیری در انسان هاست را به دست نمی دهد، اما این فرضیه که کروناویروس نوظهور از خفاش ها به وجود آمده است، بسیار محتمل می باشد. ما شواهدی را ارائه می نماییم که مطرح می کند کروناویروس نوظهور (2019-nCoV) موزائیکی نبوده و در دست کم نیمی از ژنوم خود از یک دودمان منحصر به فرد در بتاکروناویروس ها تشکیل شده است. این خصوصیات ژنومی و هم بستگی بالقوه ی آنها با ویژگی های ویروس و بیماری زایی در انسان ها درخور توجه آتی است.
خانواده ی کروناویریده شامل تعداد زیادی از ویروس هایی است که در طبیعت در پرندگان و پستانداران یافت می گردند (کان و مکاینتاش، 2005؛ فر و پرلمن، 2015 ). کروناویروس های انسانی که نخست در دهه ی 1960 توصیف شدند، در ایجاد درصد بالایی از عفونت های تنفسی هم در کودکان و هم در بزرگسالان نقش دارند (کان و مکاینتاش، 2005؛ پالز و همکاران، 2020 ).
Abstract
Background A novel coronavirus (2019-nCoV) associated with human to human transmission and severe human infection has been recently reported from the city of Wuhan in China. Our objectives were to characterize the genetic relationships of the 2019-nCoV and to search for putative recombination within the subgenus of sarbecovirus.
Methods Putative recombination was investigated by RDP4 and Simplot v3.5.1 and discordant phylogenetic clustering in individual genomic fragments was confirmed by phylogenetic analysis using maximum likelihood and Bayesian methods. Results Our analysis suggests that the 2019-nCoV although closely related to BatCoV RaTG13 sequence throughout the genome (sequence similarity 96.3%), shows discordant clustering with the Bat_SARS-like coronavirus sequences. Specifically, in the 5′-part spanning the first 11,498 nucleotides and the last 3′-part spanning 24,341–30,696 positions, 2019-nCoV and RaTG13 formed a single cluster with Bat_SARS-like coronavirus sequences, whereas in the middle region spanning the 3′-end of ORF1a, the ORF1b and almost half of the spike regions, 2019-nCoV and RaTG13 grouped in a separate distant lineage within the sarbecovirus branch.
Conclusions The levels of genetic similarity between the 2019-nCoV and RaTG13 suggest that the latter does not provide the exact variant that caused the outbreak in humans, but the hypothesis that 2019-nCoV has originated from bats is very likely. We show evidence that the novel coronavirus (2019-nCov) is not-mosaic consisting in almost half of its genome of a distinct lineage within the betacoronavirus. These genomic features and their potential association with virus characteristics and virulence in humans need further attention.
The family Coronaviridae includes a large number of viruses that in nature are found in birds and mammals (Kahn and McIntosh, 2005; Fehr and Perlman, 2015). Human coronaviruses, first characterized in the 1960s, are associated with a large percentage of respiratory infections both in children and adults (Kahn and McIntosh, 2005; Paules et al., 2020).


(جهت بزرگ نمایی روی عکس کلیک نمایید)
- اصل مقاله انگلیسی با فرمت ورد (word) با قابلیت ویرایش
- ترجمه فارسی مقاله با فرمت ورد (word) با قابلیت ویرایش، بدون آرم سایت ای ترجمه
- ترجمه فارسی مقاله با فرمت pdf، بدون آرم سایت ای ترجمه