
دانلود رایگان مقاله یک نقشه بردار ژنومی با استفاده از هم ترازی توالی مبتنی بر شاخص هیبریدی
خلاصه
زمینه: تعدادی از ابزارهای همترازسازی برای همتراز کردن ریدهای توالی یابی با ژنوم مرجع انسان ایجاد شده اند. مقیاس اطلاعات بدست امده از آزمایشات توالی یابی نسل جدید (NGS)، به سرعت در حال افزایش است. مطالعات اخیر انجام شده بر اساس فناوری NGS به طور مرتب اگزوم ها یا توالی های کامل ژنومی چندصد تا چندهزار نمونه را ایجاد کرده است. برای تامین نیاز روزافزون به آنالیز دیتاست های بسیار بزرگ NGS، لازم است که ابزارهای نقشه برداری سریع تر، حساس تر و دقیق تری ایجاد شود.
نتایج: HIA از دو شاخص جدول درهم سازی و شاخص suffix array استفاده می کند. جدول درهم سازی جستجوی مستقیم q-gram را انجام می دهد و شاخص suffix array جستجوی بسیار سریع رشته های با طول متغییر را با استفاده از جستجوی دودویی اجرا می کند. ما پی بردیم که ترکیب جدول درهم سازی و suffix array بسیار سریعتر از روش suffix array می تواند یک زیر رشته را در توالی مرجع پیدا کند. در اینجا ما منطقه ی تطابق (MR) را تعریف کردیم که طولانی ترین زیر رشته ی مشترک بین یک مرجع و یک رید است. همچنین ما مناطق همترازی کاندید (CARs) را نیز به صورت لیستی از MRs تعریف کردیم که در کنار یکدیگر قرار دارند. شاخص هیبرید برای یافتن مناطق همترازی کاندید (CARs) بین مرجع و رید استفاده شد. ما پی بردیم که همترازی نواحی بدون تطابق در CAR بسیار سریعتر از همترازی کل CAR است. در بررسی-های آزمایشی، HI در مقایسه با سایر ابزارهای همترازی نقشه برداری سریعتر و بدون کاهش چشمگیر در دقت نقشه را از خود نشان داد.
نتیجه گیری: آزمایشات ما نشان می دهد که هیبرید جدول درهم سازی و suffix array از نظر سرعت نقشه برداری ریدهای توالی یابی NGS به توالی مرجع ژنوم انسانی مفید است. در نتیجه، ابزار ما برای همتراز کردن دیتاست های عظیم به دست امده از توالی یابی NGS مناسب است.
زمینه
مطالعات اخیر براساس فناوری توالی یابی نسل جدید (NGS) صدها یا هزاران اگزوم یا توالی کامل ژنومی را با کاهش هزینه های آزمایشات NGS ایجاد کرده است. با تکامل فناوری های NGS، این فناوری ها به تدریج طول رید ها را افزایش داده و از میزان خطاها کاسته اند. برای همگام شدن با فناوری های درحال توسعه ی NGS، ابزارهای همترازسازی زیادی برای ریدهای کوتاه و بلند ایجاد شده است. این ابزارها شامل SSAHA2، BWA ، AGILE، SOAP2، Bowtie2، SeqAlto و غیره هستند. از بین اینها، بسیاری از برنامه های همترازی از راهبرد نقشه برداری مبتنی بر شاخص استفاده می کنند. برای مثال، SSAHA2، AGILE و SeqAlto از یک شاخص جدول درهم سازی (HT) یک ژنوم مرجع استفاده می کنند در حالیکه BWA، SOAP2، و Bowtie2 از یک شاخص ژنومی مبتنی بر تبدیل باروز-ویلر استفاده می کند.
تمام ابزارهای همترازی مبتنی بر HT از استراتژی seed and extended، استفاده می کنند که با جستجوی مناطق همترازی کاندید (CARs) (همترازی هر جایگاه) و گزارش بهترین همترازی ها عمل می کند. شاخص HT از جستجوی سریع جایگاه های کاندید دارای q-gram ها پشتیبانی می کند (رشته های طول q). Q کوچکتر حساسیت و تعداد CARs را افزایش می دهد اما q بزرگتر حساسیت و تعداد CARs را کاهش می دهد. علاوه بر این از آنجایی که q ثابت می شود، زمانیکه q-gram ها برای یک طول جدید نیاز باشند، HT باید مجددا ساخته شود. بیشتر ابزارهای همترازی مبتنی بر BWT از شاخص full-text minute استفاده می کنند که از نظر حافظه کارامد است و شبیه suffix tree است. از نظر زمان تطابق، suffix tree برای تطابق دقیق کارامد است اما برای تطابق غیر دقیق کند است. BWA و Bowtie2 از رویکردهای seed-and-extend مشابهی مانند استفاده از الگوریتم های مبتنی بر HT برای ریدهای طولانی پیروی می کنند.
پشتیبانی از تطابق ریدهای بلند، سرعت بالا، دقت و حساسیت، ویژگی های ضروری ابزارهای نقشه برداری NGS فعلی است. در اینجا، ما سعی کردیم مزایای استفاده از همترازی مبتنی بر HT و suffix tree در یک ابزار را بیان کنیم که این الزامات را برآورده می کند. برای این منظور، ما یک نقشه بردار ژنومی را با استفاده از همترازی توالی مبتنی بر شاخص هیبرید ایجاد کردیم.
در این مقاله، ما ابزار HIA را تشریح می کنیم، و نتایج مقایسه های انجام شده برروی کارایی این HIA و چهار ابزار همترازی محبوب دیگر شامل BWA، Bowtie2 ، SOAP2 و SeqAlto برروی داده های واقعی و شبیه-سازی شده را نشان می دهیم. نتایج تجزیه و تحلیل ازمایشی نشان می دهد که HIA در مقایسه با دیگر ابزارهای همترازی بویژه از نظر سرعت، کارامدتر است.
روش ها
شاخص هیبرید
Σ را یک الفبا و S = s0s1 … sm−1 یک رشته بر روی Σ در نظر بگیرید. |S| = m را طول S، S[i] = si را نماد iم s، S[i, j] = si … sj را یک زیر رشته و Si = S[i,m−1] یک افزونه s در نظر بگیرید. ما q-gram را به عنوان یک زیر رشته s با طول q تعریف می کنیم. در متن توالی DNA، الفبای Σ شامل چهار نوکلئوتید A، T، C و G است (به عبارتی Σ = {A, C, G, T}). ما به جای A، C، G و T به ترتیب از اعداد 0، 1، 2 و 3 استفاده می کنیم. بنابراین، هر q-gram به عنوان یک عدد صحیح بدون علامت با دو بیت در هر نوکلئوتید کدگذاری می شود. با این وجود، بسیاری از توالی های ژنومی مرجع حاوی نوکلئوتیدی غیر از این چهار نوکلئوتید مانند N هستند. این مسئله در توالی های NGS نیز اتفاق می افتد. ما N را با یک نوکلئوتید تصادفی یکنواخت مانند BWA و بسیاری از ابزارهای دیگر جایگزین می کنیم.
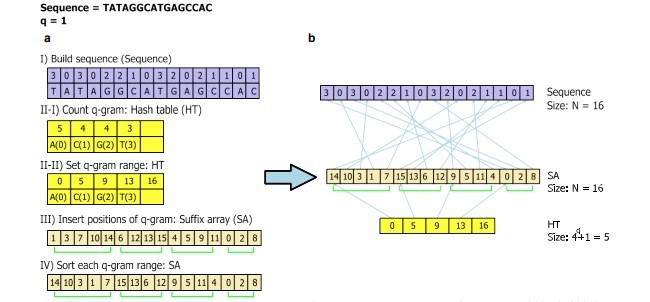
از نظر شاخص هیبرید، SOAP2 یک جدول درهم سازی را بر روی شاخص FM که یک SA فشرده است، اجرا می کند. از طرف دیگر، شاخص هیبرید دیگر حاوی یک توالی مرجع، suffix array (SA) و یک جدول درهم سازی (HT) است که در شکل 1 نشان داده شده است. به دلیل اینکه چهار نماد در این الفبا وجود دارد، توالی مرجع طول N می تواند به صورت N/4 بیت بسته بندی شود. SA یک ارایه ی موقعیت های آغازی (اعداد صحیح) افزونه های توالی مرجع در ترتیب لکسیکوگرافیکال است. تعداد افزونه های توالی اندازه ی N برابر N است. HT یک آرایه از اشاره گرها به SA است که نشان می دهد که کدام موقعیت در SA متعلق به q-gram است. از آنجا که ما یک q-gram را به عنوان یک رشته از طول q تعریف می کنیم، تعداد عناصر HT برابر 4q + 1 است. با توجه به q-gram ، HT[x] اولین موقعیت q-gram در SA است، که در آن x مقدار عددی q-gram است. ما محدوده ی (R) q-gram را در SA به صورت زیر تعریف می کنیم:
R(x) = [HT[x], HT[x + 1] − 1]
اگر q-gram در توالی وجود نداشته باشد، HT[x] اولین موقعیت q-gram بعدی در تولی است، بنابراین، HT[x] و HT[x + 1] برابر هستند و R(x) خالی است.
روش ساخت شاخص هیبرید شامل چهار فرایند است که عبارتند از : 1) بسته بندی توالی مرجع به صورت 2 بیت به ازای هر باز (توالی) ؛ 2) شمارش هر q-gram در توالی و ایجاد محدوده ی q-grams در SA (HT)؛ 3) وارد کردن موقعیت توالی هر q-gram در محدوده ی SA و 4) مرتب سازی لکسیکوگراقیکی هر q-gram (SA). بر اساس تعیین ذهنی موقعیت نهایی بسیاری از افزونه ها با استفاده از تنها چند نماد اول هر افزونه، الگوریتم ساخت SA امرتب سازی پیشوندهای طول w در افزونه های محدوده ی q-gram را انجام می دهد. اگر برخی از افزونه ها پیشوند طول W را به اشتراک بگذارند، دسته با دسته بندی طول W زیر رشته ی پیشوندهای طول W تکرار می شود. برای کاهش زمان دسترسی به توالی، پیشوندهای طول W به مقادیر صحیح تبدیل می-شوند. در صورتی که اندازه کلمه حافظه 4 بایت و اندازه حروف الفبا 4 باشد، w بین 0 تا 16 تنظیم می شود. محدوده q-gram با هم تداخل پیدا نمی کنند به طوری که فرآیند چهارم نیز می تواند به صورت موازی باشد. شکل 1 شکل 1، ساختار داده های اساسی و روش ساخت یک شاخص هیبرید برای یک توالی مرجع را توصیف می کند.
Abstract
Background: A number of alignment tools have been developed to align sequencing reads to the human reference genome. The scale of information from next-generation sequencing (NGS) experiments, however, is increasing rapidly. Recent studies based on NGS technology have routinely produced exome or whole-genome sequences from several hundreds or thousands of samples. To accommodate the increasing need of analyzing very large NGS data sets, it is necessary to develop faster, more sensitive and accurate mapping tools.
Results: HIA uses two indices, a hash table index and a suffix array index. The hash table performs direct lookup of a q-gram, and the suffix array performs very fast lookup of variable-length strings by exploiting binary search. We observed that combining hash table and suffix array (hybrid index) is much faster than the suffix array method for finding a substring in the reference sequence. Here, we defined the matching region (MR) is a longest common substring between a reference and a read. And, we also defined the candidate alignment regions (CARs) as a list of MRs that is close to each other. The hybrid index is used to find candidate alignment regions (CARs) between a reference and a read. We found that aligning only the unmatched regions in the CAR is much faster than aligning the whole CAR. In benchmark analysis, HIA outperformed in mapping speed compared with the other aligners, without significant loss of mapping accuracy.
Conclusions: Our experiments show that the hybrid of hash table and suffix array is useful in terms of speed for mapping NGS sequencing reads to the human reference genome sequence. In conclusion, our tool is appropriate for aligning massive data sets generated by NGS sequencing.
Background
Recent studies based on next-generation sequencing (NGS) technology have produced hundreds or thousands of exome or whole genome sequences with decreasing cost of NGS experiments [1]. As the NGS technologies evolve, NGS technologies have gradually increased read length and decreased error rate [2]. To keep pace with developing NGS technologies, many alignment tools have been developed for both short and long reads. These tools include SSAHA2 [3], BWA [4, 5], AGILE [6], SOAP2 [7], Bowtie2 [8], SeqAlto [9] and others. Among them, many aligning programs use index-based mapping strategy. For example, SSAHA2, AGILE and SeqAlto use a hash table (HT) index of a reference genome, whereas BWA, SOAP2 and Bowtie2 use an index of the reference genome based on the Burrows–Wheeler transform [10].
All HT-based alignment tools use the same seed-andextend strategy [11, 12], which proceeds by searching for candidate alignment regions (CARs), aligning each location, and reporting the best alignments. A HT index supports very fast lookup of candidate locations with qgrams (strings of length q). Smaller q increases the sensitivity and the number of CARs, whereas larger q decreases the sensitivity and number of CARs. Furthermore, because q is fixed, the HT must be rebuilt when q-grams of a new length are needed. Most BWT-based alignment tools use the full-text minute index [13], which is memory-efficient and similar to the suffix tree. With respect to matching time, the suffix tree is efficient for exact matching, although it is slow for inexact matching. BWA and Bowtie2 follow similar seed-and-extend approaches, including the use of HT-based algorithms for long reads.
Support of long-read alignment, high speed, accuracy, and sensitivity are essential features of the current NGS mapping tools. Here, we tried to merge the advantages of HT-based and suffix tree-based alignment in a tool that satisfies these requirements. To this end, we developed a genome mapper using hybrid index-based sequence alignment (HIA).
In this article, we describe the HIA tool, and show the results of comparisons of performance on simulated and real read data between HIA and the other four popular alignment tools including BWA, Bowtie2, SOAP2 and SeqAlto. The results of the benchmark analysis demonstrate that HIA outperforms the other aligners, especially inspeed.
Methods
Hybrid index
Let Σ be an alphabet and S = s0 s1 … sm−1 be a string over Σ. Let |S| = m be the length of S, S[i] = si be the i-th symbol of S, S[i, j] = si … sj be a substring, and Si = S[i,m−1] be a suffix of S. We define a q-gram as a substring of S with length q. In the context of DNA sequence, the alphabet Σ consists of the four nucleotides A, C, G, and T, i.e., Σ = {A, C, G, T}. We assign A, C, G, and T to the numbers 0, 1, 2, and 3, respectively. Thus, each q-gram is encoded as an unsigned integer with two bits per nucleotide. However, most reference genome sequences contain a nucleotide other than {A, C, G, T}, such as ‘N’. This occasionally happens with NGS read sequences as well. We replace ‘N’ with a uniform random nucleotide such as BWA and many other tools do.
In terms of a hybrid index, SOAP2 implements a hash table on the FM-index which is a compressed SA. On the other hand, our hybrid index consists of a reference sequence, a suffix array (SA), and a hash table (HT), as in Fig. 1. Since there are four symbols in the alphabet, a reference sequence of length N can be packed into N/4 bytes. The SA is an array of the starting positions (integers) of suffixes of the reference sequence in lexicographical order. The number of suffixes of a sequence of size N is N. The HT is an array of pointers into SA indicating which positions in SA belong to which q-grams. Since we define a q-gram as a string of length q, the number of elements of HT is 4 q + 1. Given a q-gram, HT[x] is the first position of the q-gram in SA, where x is the numeric value of the q-gram. We define the range (R) of the q-graminSAby (1):
R(x) = [HT[x], HT[x + 1] − 1]
If the q-gram does not exist in the sequence, HT[x] is the first position of next existing q-gram in the sequence so that HT[x] and HT[x + 1] are the same, and R(x) is empty.
The procedure for constructing the hybrid index consists of four processes, as follows: (1) packing the reference sequence in a 2-bits per base format (Sequence); (2) counting each q-gram in the sequence and set the range of q-grams in SA (HT); (3) inserting the sequence positions of each q-gram into its range of SA; and (4) sorting the range for each q-gram (SA) lexicographically. Based on the heuristic determining the final positions of most of the suffixes using only the first few symbols of each suffix [14], the SA construction algorithm proceeds sorting the length-w prefixes of the suffixes in a q-gram range. If some suffixes share also a length-w prefix, then the sort is repeated by sorting the length-w substrings that follow the length-w prefixes, and so on. In order to reduce the sequence access time, length-w prefixes are converted to integer values. In the case that the size of the memory word is 4 bytes and the size of the alphabet is 4, the w is set to between 0 and 16. The ranges of q-grams are not overlapped so that the fourth process can also be parallelized. Figure 1 describes the underlying data structure and the method for constructing a hybrid index of a reference sequence.
خلاصه
زمینه
روش ها
شاخص هیبرید
نقشه برداری هیبرید: یافتن نواحی همترازی کاندید (CAR)
نقشه برداری هیبرید : همترازی ناحیه ی همترازی کاندید (CAR)
نقشه برداری هیبرید ممتد
اجرا
نتایج و بحث
ارزیابی مجموع داده ها و معیارهای ارزیابی
نتایج ارزیابی
عملکرد ایجاد شاخص
نتایج ریدهای شبیه سازی شده ی single‑end
نتایج ریدهای شبیه سازی شده ی paired‑end
نتایج مجموع داده های واقعی
نتایج تست های چند رشته ای
نتیجه گیری
منابع
Abstract
Background
Methods
Hybrid index
Hybrid mapping: finding candidate alignment region (CAR)
Hybrid mapping: aligning candidate alignment region (CAR)
Extended hybrid mapping
Implementation
Results and discussion
Evaluationdatasets andevaluationmeasurements
Evaluation results
Performance of index generation
Results of simulated single end reads
Results of simulated paired end reads
Results of real datasets
Results of multithreading tests
Conclusions
References