
دانلود رایگان مقاله تکامل همگرا از عملکرد مشابه در دو آنزیم ساختاری متفاوت
یک نمونه از دو آنزیم مرتبط به هم که واکنش های مشابهی را کاتالیز می کنند اما مکان های فعال مختلفی نسبت به هم دارند، با مقایسه کردن ساختار آنزیم کاهش دهنده تیوردوکسین اشیرشیا کولی با آنزیم کاهش دهنده گلوتاتیون، ارائه شده است. هر دوی این آنزیم ها، آنزیم های دیمری هستند که باعث کاتالیز شدن کاهش دی سولفید ها توسط هسته های پیریدین از طریق یک دی سولفید آنزیم و یک فلاوین، می شود. آنزیم های کاهش دهنده گلوتاتیون در انسان شامل چهار دامنه ساختاری در هر مولکول هستند: دو هسته ای های فلاوین- آدنین (FAD)- و دامنه های اتصالی به نیکوتینامید- آدنین دو هسته ای فسفات (NADPH) ، دامنه مرکزی و دامنه پایانه C که باعث می شود یک سطح مشترک دیمری و بخشی از مکان فعال ایجاد شود. با وجود این که هر دوی این آنزیم ها مکانیزم های کاتالیزی و ساختار های سه گانه مشابه با هم دارند، مکان های فعال آن ها شبیه هم نیست. ما ساختار کریستالی اشیرشیا کولی تیوردوکسین را با تفکیک 2A مشخص کرده ایم و نشان دادیم که آنزیم کاهش دهنده تیوردوکسین ، دامنه ای که باعث شکل گیری سطح مشترک دیمری در آنزیم کاهش دهنده گلوتاتیون می شود را ندارد و کاملا ساختار دیمری متفاوتی را از خودش نشان میدهد. دی سولفید های فعال از نظر کاتالیزی نیز در دامنه های متفاوتی در طرف های مخالف در سیستم حلقه فلاوین قرار دارند. این موضوع نشان میدهد که این آنزیم ها از پروتئین های پیش زمینه ای هسته ای متفاوتی شکل گرفته اند و مستقل از هم دیگر، فعالیت های آنزیم کاهش دهنده دی سولفید را ایفا می کنند.
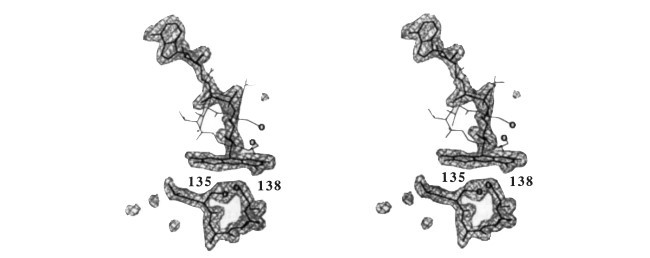
تیوردوکسین ها پروتئین های فعال اکسایشی کوچکی هستند که عملکرد های مختلفی را ایفا میکنند. در ای کولی ( اشیرشیا کولی) تیوردوکسین ها توسط آنزیم کاهش دهنده تیوردوکسین مبتنی بر NADPH کاهش پیدا می کنند. ساختار سه بعدی از TR بر اساس جایگزینی های مختلف هم شکل ایجاد می شود ( شکل1) و شامل دو مولکول می باشد که به صورت بسیار نزدیک با یکدیگر تعامل دارند که باعث شکل گیری دیمر های متقارن می شود. هر کدام از مولکول ها دارای سه دامنه معین می باشد که متناظر با FAD، NADPH و دامنه های مرکزی از آنزیم کاهش دهنده گلوتاتیون (GR) ( شکل 2) می باشند. ساختار سه بعدی از TR بسیار شبیه به سه دامنه اول از GR می باشد و تفاوت اصلی بین آن ها، تغییر در جهت دامنه NADPH و حذف شدن یک هلیکس می باشد (شکل2). نوزده مورد از رشته های 21 بتا در TR متناظر با المان های GR هستند؛ دو مورد دیگر در پایانه T و نزدیک به دی سولفید فعال اکسایشی قرار دارد که ویژگی متمایز در توالی TR ها می باشد (شکل3). به صورت مشابه، پنج مورد از هفت مورد آلفا هلیکس ها و سه مورد از هلیکس های 3t0 در TR مطابق با المان های مشابه در GR هستند. یک ویژگی بسیار مهم از تنظیم ساختاری، جابجایی های نسبی از دامنه های NADPH در دو آنزیم می باشد. این موضوع مطابق با چرخش یکی از این دامنه ها به مقدار 66 درجه حول رشته های بتا می باشد که به FAD و دامنه مرکزی متصل می شوند و باعث می شوند که ساختار پایه نسبت بدون تغییر بماند ( شکل 2). در تنظیم دامنه های FAD / مرکزی و تفکیک 2A برای دامنه های NADPH برای پس ماند های اصلی ، باعث می شود که 70% از توالی TR شکل بگیرد از جمله تنظیم اتم های آلفا هلیکس که انحراف کمتر از 2.5A دارند و باعث شکل گیری انحراف موثر 1.1 A هم در دامنه های مرکزی FAD و مرکزی و هم در دامنه های NADPH می شود و 54% و 65% از پس ماند های TR بر اساس این معیار در دو بخش از ساختار، صدق می کنند. این نتایج مشابه با انحراف های شناسایی شده بر روی ساختار های هم پوشان گلوبین با شباهت توالی نسبی می باشد.
سیستئین های فعال اکسایشی در مکان های مختلف از این دو آنزیم قرار گرفته اند ( شکل 1 و 2). اما هر دوی آنزیم ها دارای شباهت های توالی یا ساختاری در منطقه های مرتبط با دی سولفید دیگر آنزیم ها می باشند توالی TSDGFF ( نماد های آمینو اسید ها با یک حرف) در GR ( پس ماند 176-181) مطابق با توالی مکان فعال TCDGFF در TR می باشد و این هگزاپپتید، یکی از بزرگترین شباهت های توالی در کل این تراز می باشد ( شکل 3). در مکان دی سولفید های GR، ( پس ماند 58-63)، اولین دور از آلفا هلیکس در تراز ساختاری با هلیکس 3t0 در TR قرار دارد ( شکل 2 و 3). همراه با هماهنگی در المان های ساختاری ثانویه و حضور چند ناحیه خوشه ای دیگر با شباهت توالی بالا، این موضوع نشان میدهد که هر دوی این پروتئین ها از یک پیشینه مشترک ایجاد شده اند. دامنه های FAD و NADPH از TR و GR هر دو شامل موتیف های اتصالی به هسته و یک صفحه غیر موازی بتا اضافی هستند که ممکن است در اثر تغییرات ژنی ایجاد شده باشد. مقایسه های قبلی در رابطه با GR و مونو اکسیژنا شامل FAD، یعنی هیدروکسیلاز هیدروکسی بنزوئات p نشان دهنده شباهت های ساختاری در دامنه های FAD و بعضی دیگر از المان های مشترک در دامنه مرکزی می باشد. المان های متفاوت همراه با المان های مشابه در این مطالعه شناسایی شده اند اما تمایز بین دو مکانیزم دشوار می باشد که این موضوع به دلیل غیاب شباهت توالی ها ، کمبود شباهت ساختاری در NADPH و تفاوت های بنیادی در واکنش هایی می باشد که این آنزیم ها موجب کاتالیز آن ها می شوند. در مقابل، کاتالیز های TR و GR دارای واکنش های بسیار مشابه می باشند که تنها از نظر بستر متفاوت هستند و شباهت های توالی آن ها نشان دهنده رابطه بین این سه دامنه از TR می باشد.
AN example of two related enzymes that catalyse similar reactions but possess different active sites is provided by comparing the structure of Escherichia coli thioredoxin reductase with glutathione reductase 1• Both are dimeric enzymes that catalyse the reduction of disulphides by yridine nucleotides through an enzyme disul phide and a ftavin . Human glutathione reductase contains four structural domains within each molecule: the ftavin-adenine di nucleotide (FAD)- and nicotinamide-adenine dinucleotide phos phate (NADPH)-binding domains, the 'central' domain and the C-terminal domain that provides the dimer interface and part of the active site3.4. Although both enzymes share the same catalytic mechanism and similar tertiary structures, their active sites do not resemble each other5•6• We have determined the crystal structure of E. coli thioredoxin reductase at 2 A resolution, and show that thioredoxin reductase lacks the domain that provides the dimer interface in glutathione reductase, and forms a completely different dimeric structure. The catalytically active disulphides are located in different domains on opposite sides of the ftavin ring system. This suggests that these enzymes diverged from an ancestral nucleotide-binding protein and acquired their disulphide reductase activities independently.
Thioredoxins are small redox-active proteins that have diverse functions. In E. coli, thioredoxin is reduced by NADPH-depen dent thioredoxin reductase (TR; ref. 7)_ The three-dimensional structure of TR was determined by multiple isomorphous replacement (Fig. 1) and consists of two molecules which inter act closely to form a symmetric dimer. Each molecule has three clearly delineated domains, which correspond to the FAD, NADPH and central domains of glutathione reductase (GR) (Fig. 2). The tertiary structure of TR is very similar to that of the first three domains of GR, the chief differences being a change in the orientation of the NADPH domain and the dele tion of a helix (Fig. 2). Nineteen of the 21 f3 strands in TR correspond to elements in GR; the other two are located at the N terminus and near the redox-active disulphide that is a distinct feature of the TR sequence (Fig. 3). Likewise, five of the seven a helices and three of the four 310 helices in TR align with corresponding elements in GR. A notable feature of the struc tural alignment is the relative displacement of the NADPH domains in the two enzymes. This corresponds to a rotation of one of these domains by 66° about the two f3 strands that connect it to the FAD and central domains, leaving the backbone structure relatively unperturbed (Fig. 2). On aligning the FAD/central domains and the NADPH domain separately, the root-mean-square (r.m.s.) deviation in Ca positions is 1.8 A for the FAD/ central domains and 2.0 A for the NADPH domain for core residues comprising about 70% of the TR sequence. Including in the alignment only those Ca atoms that deviate by less than 2.5 A results in r.m.s. deviations of 1.1 A both in the FAD/central domains and in the NADPH domain, with 54% and 65% of the TR residues satisfying this criterion in the two parts of the structure, respectively . These results are similar to the deviations obtained on superimposing globin structures with comparable sequence identity8.
The redox-active cysteines are in different locations in the two enzymes (Figs 1 and 2). But, both enzymes have sequence or structural similarities in the regions corresponding to the disulphide of the other enzyme. The sequence TSDGFF (single letter amino-acid notation) in GR (residues 176-181) aligns with the active-site sequence TCDGFF in TR, and this hexapeptide is the one with greatest sequence similarity in the entire align ment (Fig. 3). At the site of the GR disulphide (residues 58-63), the first turn of the a helix is in structural alignment with a 310 helix in TR (Figs 2 and 3). Together with the homology in secondary structural elements and the presence of several other clustered regions of relatively high sequence similarity, this suggests that both proteins have evolved from a common ances tor . The FAD and NAO PH domains of TR and GR both contain the f3af3 nucleotide-binding motif 9 and an additional anti parallel f3 sheet, and may have arisen by gene duplication 10 Previous comparison of GR and an FAD-containing monoxy genase, p-hydroxybenzoate hydroxylase, revealed structural similarity in their FAD domains and some common elements in their central domains 11• Elements of divergent as well as convergent evolution were indicated, but distinguishing between the two mechanisms was difficult because of the absence of sequence identity, the lack of structural similarity in the NAO PH domains and fundamental differences in the reactions cata lysed 11. By contrast, TR and GR catalyse very similar reactions that differ only in substrate, and share a sequence similarity showing relationships in all three domains of TR6.