
دانلود رایگان مقاله گام های بزرگ در تعیین توالی DNA
در سال های اخیر پیشرفت های فوق العاده ای در توانایی ما برای تعیین سریع و کم هزینه توالی DNA توالی به وجود آمده است. این مساله تحولات بنیادین در زمینه های ژنتیک و زیست شناسی ایجاد کرده که منجر به درک عمیق تر از رویدادهای مولکولی در فرایندهای زیستی شده است. پیشرفت های سریع تکنولوژیکی، فرصت ها و کاربردهای تعیین توالی را به سرعت گسترش داده است و در عین حال چالش هایی در مراحل پیشین تعیین توالی و در فرآیندهای پایین دستی بررسی و تجزیه و تحلیل این حجم انبوه از داده های توالی تحمیل کرده است. به طور سنتی، تعیین توالی، به قطعات کوچک DNA برای حدود یک هزار پایه(مشتق شده از ژنوم ارگانیسم) بمنظور حفظ کیفیت توالی و دقت بالا برای خواندن طول های بالاتر محدود شده است. اگر چه پیشرفت های تکنولوژیکی بسیاری بوجود آمده است، روش های تعیین توالی موازی که بطور تجاری در حال حاضر در دسترس هستند قادر به حل این مساله نیستند. روش تعیین توالی قادر به حل این مسئله نبوده است. با این حال، اعلان اخیر در تعیین توالی نانوحفره وعده از بین بردن این محدودیت خواندن طول را داده است، که امکان تعیین توالی قطعات سالم بزرگ تر DNA را می دهد. توانایی تعیین توالی DNA سالم با دقت بالا گامی بزرگ به سوی تسهیل تجزیه و تحلیل پایین دستی و افزایش قدرت تعیین توالی نسبت به امروز است. این بررسی برخی از پیشرفت های فنی در تعیین توالی که مرزهای جدیدی را در ژنتیک باز کرده-اند پوشش می دهد.
1. مقدمه
درک کامل زبان DNA نیاز به تعیین کامل ترتیب پایه های ژنوم انسان (و یا دیگر موجودات مورد نظر) دارد. دستیابی به آن دانش، وعده بینش کامل تر نسبت به تغییرات بیولوژیکی و علل بیماری ها را می دهد. در سپیده دم تعیین توالی، خواندن ترتیب چهار پایه DNA یک فرایند دست و پا گیر بود. تعیین توالی اگرچه هنوز به سختی، با معرفی روش تجزیه شیمیایی صنعتی توسط Maxam و Gilbert [1] و روش تعیین توالی زنجیره پایانی که توسط sanger ساخته شده بود[2] در پایان دهه 1970 ممکن شد. ثابت شد روش دوم مفید تر است و روش غالب تعیین توالی DNA برای تقریبا سه دهه بود و پروژه ژنوم انسان (HGP) را به پیش برد، و هنوز هم توسط بسیاری به عنوان "استاندارد طلایی" در نظر گرفته می شود. راه اندازی تجاری ابزارهای تعیین توالی موازی فشرده DNA در سال 2005 آغاز تغییر پارادایم طراحی شده توسط تکنیک-های جدید تعیین توالی DNA بود که به محققان برای پاسخ به سوالات جسورانه در آزمایشات وسیع ژنوم الهام بخشید. در سال های اخیر رقبای جدید بسیاری در زمینه تعیین توالی موازی فشرده دیده می شود. قابل ذکر است، روش های تعیین توالی نوآورانه با استفاده از تک مولکول های DNA و تشخیص بی درنگ ظهور کرده اند. در حال حاضر، این رویکردهای نواورانه در حال تکمیل پلتفرم های تعیین توالی موجود است، اما پیش از آن، آنها مسیری طولانی برای جایگزین شدن با روش تعیین توالی موازی فشرده دارند.
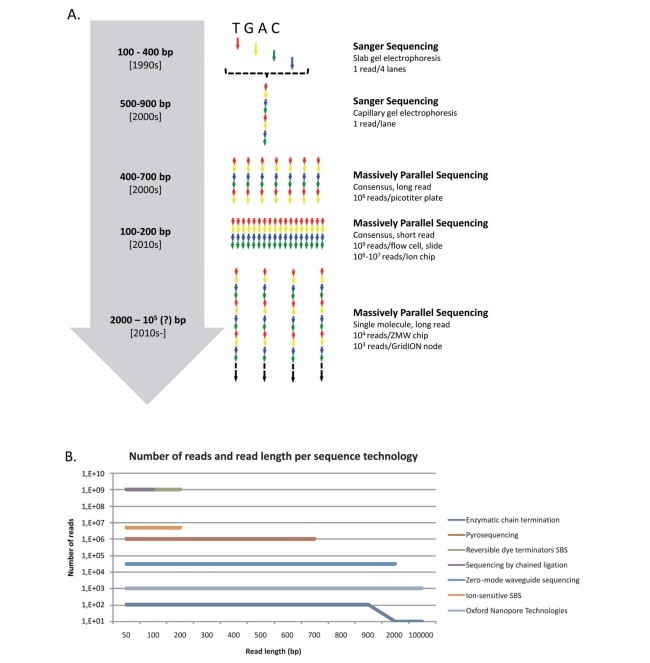
فن آوری های معمول تعیین توالی DNA و چالش های خود و محدودیت هایشان، در زیر توضیح داده می-شود. خلاصه ای از ویژگی های هر یک از تکنولوژی ها در جدول 1 ارائه شده است حالی که تعدادی از خوانش توالی ها و خوانش طول در شکل 1 نشان داده شده است.
2. توالی sanger – توالی خاتمه دهی زنجیره
مهارت روش خاتمه دهی زنجیره در استفاده از نوکلئوتیدهای خاتمه زنجیره، دیدئوکسی- نوکلئوتید است که فاقد یک گروه 3پریم هیدروکسیل است و بسط و گسترش بیشتر زنجیره DNA کپی شده را محدود می-کند. برای تعیین توالی اولیه sanger نیاز است فرآیند توالی به چهار واکنش جداگانه تقسیم شود. هر واکنش شامل یک الگوی تک رشته ای ،DNA یک آغازگر DNA و یک پلیمراز DNA در حضور مخلوطی از چهار نوکلئوتید اصلاح نشده، که یکی از آنها نشانه گذاری شده، و یک نوع نوکلئوتید خاتمه زنجیره اصلاح شده است. قطعاتی با طول متفاوت پس از هیبریداسیون پرایمر و اکتنشن پلیمراز سنتز می شوند و همه همان 5 پریمپایانی را دارند اما توسط یک نوکلئوتید خاتمه زنجیره به 3ختم می شوند. اضافه کردن تنها بخشی از نوکلئوتید پایان دار ترکیب تصادفی دیدئوکسی نوکلئوتیدها را تنها در بخش کوچکی از مولکول ها تضمین می-کند. از آنجا که نوکلئوتیدهای خاتمه زنجیره مختلف در چهار واکنش توالی یابی استفاده می شود، تمام ترکیبات خاتمه می تواند تولید شود. الگوهای DNA محدود به 3 تولید شده پس از تغییر ماهیت بوسیله حرارت و تجزیه و تفکیک شدن توسط ژل الکتروفورز، فراورده های هر چهار واکنش تعیین توالی موازی را ادامه می دهد[2]. با استفاده از رادیواکتیو یا، اخیرا، بوسیله برچسب زنی با فلورسنت برای تجسم باندها دنباله -ای از الگوهای DNA اصلی را قادر می سازد تا با پیروی از ترتیب مهاجرت قطعات بزرگتر متوالی در ژل تعیین شود.
چند پیشرفت در روش اصلی توسعه یافته توسط sanger بوجود آمده است. اینها عبارتند از: برچسب زنی خاتمه زنجیره نوکلئوتیدها با رنگهای فلورسنت طیفی مجزا که قادر می سازد: هر لوله و خط تنها در گام های تولید و شکستن قطعه مورد استفاده قرار گیرد [3، 4]؛ حذف نیاز به کست ژل با استفاده از الکتروفورز ژل مویرگی [5، 6]؛ و اتوماسیون پروتکل، که منجر به افزایش موازی سازی، تکرارپذیری و توان عملیاتی می-شود[7]. توالی sanger هنوز به طور گسترده ای امروز برای بسیاری از کاربردها، به خصوص اعتبارسنجی متغیرهای ژنتیکی و در مواردی که کیفیت بالای خوانش 300-900 پایه مورد نیاز است استفاده می شود. با این حال، پیشرفت های عمده ای در تکنولوژی توالی در سال های اخیر به روش توالی sanger منتسب نشده است، اما روش های تعیین توالی موازی فشرده به سرعت در حال پدیدار شدن است.
3. توالی موازی فشرده - توالی اجماع
پلتفرم های تعیین توالی که امروزه به طور گسترده ای در تحقیقات ژنتیکی مورد استفاده قرار می گیرند، پلتفرم-های تعیین توالی فشرده هستند. که ویژگی های بسیاری از توالی sanger به ارث برده اند مانند استفاده از پلیمرازها برای سنتز، نوکلئوتیدهای اصلاح شده و تشخیص فلورسنت. ویژگی دیگر این است که آنها نیاز به DNA دارند تا بطور کلونال تقویت شوند تا تشکیل یک الگوی اجماعی قبل از تعیین توالی را بدهند.
این روش های تعیین توالی به نام نسل بعدی تعیین توالی خوانده می شوند، روش های تعیین توالی با توان عملیاتی بالا، و یا نسل دوم تعیین توالی. با این حال، همچنانکه تکنولوژی های تعیین توالی توسعه می یابند، حفظ رجوع به فناوری های تعیین توالی قدیمی و بدیع در حال ظهور متعلق به یک نسل خاص سخت است. بنابراین، بهتر است آنها را بر حسب برجسته ترین ویژگی های مشترکشان ، مانند روش های تعیین توالی موازی فشرده و یا تک مولکولی گروه بندی کنیم. در حال حاضر، پنج رقیب در زمینه فناوری های تعیین توالی موازی فشرده وجود دارد، هر کدام با نقاط قوت و ضعف خاص. این ها در ادامه توصیف و مورد بحث قرار گرفته اند.
3.1. تعیین توالی به روش پیرو
Melamede در ابتدا مفهوم تعیین توالی با سنتز (SBS) را در سال 1985 در یک گزارش از تلاش ها برای تشخیص رویداد ترکیب نوکلئوتید با اندازه گیری جذب نوکلئوتید [8] را مطرح کرد. بی خبر از یافته های Melamede ، Nyrén رویکرد SBS دیگری را با استفاده از زیست تابی به جای جذب در سال 1986 تصور کرد[9]، که منجر ( 12 سال پس از آزمایش دیگری) به معرفی pyro-sequencing شد[10]. فن-آوری pyrosequencing قادر به تکمیل توالی sanger بود و بیشتر توسط علوم زندگی 454، که توسط جاناتان روتبرگ پایه گذاری شد، توسعه داده شد. در سال 2005، روتبرگ و همکارانش [11] اولین مقاله اثبات مفهومی را منتشر کردند که نشان داد رویکرد pyrosequencing در تعیین توالی موازی فشرده، بازده فوق العاده ای در افزایش ظرفیت توالی، و در نتیجه دگرگونی روش تعیین توالی دارد.
در SBS، این رویداد است که تشخیص داده می شود ترکیب نوکلئوتید با رشته های DNA در حال رشد است. همچنانکه نوکلئوتیدها توسط پلیمراز ترکیب می شوند، پیرو فسفات (PPI) و پروتون ها تولید می شوند. در pyrosequencing، PPI در یک آبشار آنزیمی برای تولید یک انفجار نوری مورد استفاده قرار می گیرد. PPI بوسیله سولفوریلاز ATP به ATP تبدیل می شود، که سپس از آن برای تولید فوتون توسط لوسیفراز انزیم کرم شب تاب آنزیم استفاده می شود، که یک سیگنال نوری متناسب با تعداد نوکلئوتیدهایی که گنجانیده شده است فراهم می کند. چالش، تشخیص چشمک های نور از هر الگوی DNA منحصر به فرد است هنگام تعیین توالی الگوهای متعدد به صورت موازی است. انفصال فضایی هر واکنش توالی بر روی مهره های ته نشین شده در ته چاه کوچک بر روی یک صفحه picotiter به زیبایی این مشکل را حل کرد.
pyrosequencing موازی فشرده با قطعه قطعه شدن DNA و اتصال آداپتوری آغاز می شود. الگوهای تک رشته ای DNA سپس بر روی دانه ها محدود می شوند و امولسیون PCR انجام می شود، بطور کلونالی هر الگوی DNA در میکروراکتورهای آبی جدا شده با روغن تقویت می شود. امولسیون سپس شکسته می شود و دانه های حامل DNA از دانه های خالی در فرآیندی به نام غنی سازی از هم جدا می شوند. دانه های غنی در چاه های کوچک در صفحه picotiter با پرایمر و پلیمراز DNA پلیمراز با هم ته نشین می شوند. در حالت ایده آل، تنها یکی از این دانه ها برای هر چاه مناسب خواهد بود. دانه های کوچکتر نیز اضافه می شوند، که آنزیم های مسئول تولید نور با استفاده از PPI را حمل می کنند. نوکلئوتیدها پس از آن از روی زیر لایه در یک جریان آرام از محلول مورد استفاده در یک ترتیب از پیش تعیین شده عبور کرد، و چشمک های نور در هر چاه متناظر با ترکیب ثبت شد. حذف کارآمد واکنش با محصولات، که در صورت عدم حذف می تواند واکنش توالی را آشفته کند، توسط جریان آرام تسهیل می شود. از این رو، الگوهای توالی DNA با اگاهی از محل آنها تعیین می شود، منظور جریان نوکلئوتید و سوابق هر یک از فلش های نور از هر چاه است[10-12]. اشکال عمده استفاده از این رویکرد، مشکلات در بسط توالی نوکلئوتیدهای یکسان (مناطق homopolymeric) طولانی تر از حدود پنج نوکلئوتید با توجه به پاسخ غیرخطی نوری است که آن ها تولید می کنند[10].
In recent years there have been tremendous advances in our ability to rapidly and cost-effectively sequence DNA. This has revolutionized the fields of genetics and biology, leading to a deeper understanding of the molecular events in life processes. The rapid technological advances have enormously expanded sequencing opportunities and applications, but also imposed strains and challenges on steps prior to sequencing and in the downstream process of handling and analysis of these massive amounts of sequence data. Traditionally, sequencing has been limited to small DNA fragments of approximately one thousand bases (derived from the organism’s genome) due to issues in maintaining a high sequence quality and accuracy for longer read lengths. Although many technological breakthroughs have been made, currently the commercially available massively parallel sequencing methods have not been able to resolve this issue. However, recent announcements in nanopore sequencing hold the promise of removing this read-length limitation, enabling sequencing of larger intact DNA fragments. The ability to sequence longer intact DNA with high accuracy is a major stepping stone towards greatly simplifying the downstream analysis and increasing the power of sequencing compared to today. This review covers some of the technical advances in sequencing that have opened up new frontiers in genomics.
1 Introduction
Fully understanding the language of DNA requires the complete determination of the order of bases in the genome of humans (or other organisms of interest). Acquiring that knowledge promises to yield more complete insights into biological variations and etiology of diseases. In the dawn of sequencing, reading the order of the four bases of DNA was a cumbersome process. Sequencing was made possible, although still difficult, by the introduction of the chemical degradation method of Maxam and Gilbert [1] and Sanger’s chain-termination sequencing method [2] at the end of the 1970s. The latter proved to be more useful and was to be the dominant DNA sequencing technique for almost three decades, propelled by the Human Genome Project (HGP), and is still considered by many the “gold standard”. The commercial launch of massively parallel DNA sequencing instruments in 2005 initiated a paradigm shift powered by new DNA sequencing techniques that inspired researchers to address bolder questions in genome-wide experiments. Recent years have seen many new contenders in the field of massively parallel sequencing. Notably, innovative novel sequencing methods using single molecules of DNA and real-time detection have emerged. Currently, these novel approaches are complementing existing sequencing platforms, but they have a long way to go before replacing massively parallel sequencing.
The most commonly used DNA sequencing technologies, and their challenges and limitations, are described below. A summary table of the features of each technology is provided in Table 1 while the number of sequence reads and read lengths are shown in Fig. 1.
2 Sanger sequencing – chain-termination sequencing
The ingenuity of the chain-termination technique lies in the use of chain-terminating nucleotides, dideoxy - nucleotides that lack a 3’-hydroxyl group, restricting further extension of the copied DNA chain. Early Sanger sequencing required the sequencing process to be split into four separate reactions. Each reaction involves a singlestranded DNA template, a DNA primer and a DNA polymerase in the presence of a mixture of the four unmodified nucleotides, one of which is labeled, and a type of modified chain-terminating nucleotide. Fragments of varying length are synthesized after primer hybridization and polymerase extension, all having the same 5’ end but terminated by a chain-terminating nucleotide at the 3’ end. Adding only a fraction of the terminating nucleotide ensures the random incorporation of dideoxynucleotides in only a small subset of molecules. Since different chain- terminating nucleotides are used in the four sequencing reactions, all combinations of termination can be produced. The generated 3’-terminated DNA templates are then heat-denatured and fractionated by gel-electro pho - resis, running products of all four sequencing reactions in parallel [2]. Using radioactive or, more recently, fluorescent labeling to visualize the bands enables the sequence of the original DNA template to be determined by following the migration order of successively larger fragments in the gel.
Several enhancements have been made to the original method developed by Sanger. These include: labeling the chain-terminating nucleotides with spectrally distinct fluorescent dyes enabling: a single tube and lane to be used in the fragment generation and fractionation steps, respectively [3, 4]; elimination of the need to cast gels by using capillary gel electrophoresis [5, 6]; and automation of the protocol, leading to increases in parallelization, reproducibility and throughputs [7]. Sanger sequencing is still widely used today for many applications, particularly validation of genetic variants and in cases where high quality reads of 300–900 bases are needed. However, the major advances in sequencing technology in recent years have not been related to the mature Sanger sequencing method, but to the rapidly evolving massively parallel sequencing methods.
3 Massively parallel sequencing – consensus sequencing
The most widely used sequencing platforms in genetic research today are the massively parallel sequencing platforms, which have inherited many features from Sanger sequencing, such as the use of polymerases for synthesis, modified nucleotides and fluorescent detection. Another feature is that they require the DNA to be clonally amplified forming a consensus template prior to sequencing.
These sequencing methods have been called next generation sequencing, high-throughput sequencing methods, or second-generation sequencing. However, as the sequencing technologies continue to develop, it is hard to keep referring to old and novel emerging sequencing technologies as belonging to a certain generation. Therefore, it is preferable to categorize them by their most prominent common feature, such as being massively parallel or single-molecule sequencing methods. Currently, there are five competing massively parallel sequencing technologies, each with specific strengths and weaknesses. These are described and discussed below.
3.1 Pyrosequencing
Melamede originally outlined the concept of sequencingby-synthesis (SBS) in 1985, in a report of efforts to detect nucleotide incorporation events by measuring nucleotide absorbance [8]. Unaware of Melamede’s findings, Nyrén conceived another SBS approach using bioluminescence instead of absorbance in 1986 [9], which led (after another 12 years of experiments) to the introduction of pyro - sequencing [10]. Pyrosequencing technology was able to complement Sanger sequencing and was further developed by 454 Life Sciences, founded by Jonathan Rothberg. In 2005, Rothberg and colleagues [11] released the first proof-of-concept paper demonstrating a massively parallel pyrosequencing approach, yielding a tremendous increase in sequence capacity, and thereby transforming the way sequencing was conducted.
In SBS, the event that is detected is the incorporation of nucleotides into growing DNA strands. As the nucleotides are incorporated by the polymerase, pyrophosphate (PPi) and protons are generated. In pyrosequencing, the PPi is used in an enzymatic cascade to generate a light burst. The PPi is converted by ATP sulfurylase to ATP, which is then used by the firefly enzyme luciferase to generate photons, providing a light signal that is proportional to the number of nucleotides being incorporated. The challenge is to detect the flashes of light from each unique DNA template while sequencing numerous templates in parallel. Spatially separating each sequencing reaction on beads deposited in small wells on a pico - titer plate elegantly solved this problem.
Massively parallel pyrosequencing begins with fragmentation of the DNA and adaptor ligation. Single-stranded DNA templates are then bound on beads and emulsion PCR is performed, clonally amplifying each DNA template in aqueous microreactors isolated by oil. The emulsion is then broken and the beads carrying DNA are separated from empty beads in a process called enrichment. The enriched beads are deposited in the small wells on the picotiter plate together with primer and DNA polymerase. Ideally, only one of these beads will fit into each well. Smaller beads are also added, carrying the enzymes responsible for the light generation using PPi. Nucleotides are then passed over the substrate in a laminar flow of solutions applied in a predetermined order, and the flashes of light in each well representing incorporation events are recorded. Efficient removal of reaction by-products, which could otherwise perturb the sequencing reaction, is facilitated by the laminar flow. Hence, the sequence of the DNA templates is determined from the knowledge of their location, the order of the flow of nucleotides and records of each flash of light from each well [10–12]. The major drawback using this approach are difficulties in sequencing stretches of identical nucleotides (homopolymeric regions) longer than approximately five nucleotides due to the nonlinear light response they generate [10].
1. مقدمه
2. توالی sanger – توالی خاتمه دهی زنجیره
3. توالی موازی فشرده - توالی اجماع
3.1. تعیین توالی به روش پیرو
3.2.SBS نابودگر رنگ برگشت پذیر
3.3. توالی بوسیله زنجیره لیگاسیون
3.4. توالی با استفاده از لیگاسیون حذف زنجیره
SBS 3.5. حساس یونی
3.6. موضوعات جاری در تعیین توالی موازی فشرده
4. تعیین توالی موازی فشرده – تعیین توالی تک مولکولی
SBS 4.1. ترمیناتور تک رنگ برگشت پذیر
4.2. تعیین توالی موجبر حالت صفر با پلیمراز تثبیت شده
4.3. تعیین توالی نانوحفره
5. چشم انداز آینده
منابع
1 Introduction
2 Sanger sequencing – chain-termination sequencing
3 Massively parallel sequencing – consensus sequencing
3.1 Pyrosequencing
3.2 Reversible dye terminator SBS
3.3 Sequencing by chained ligation
3.4 Sequencing by unchained ligation
3.5 Ion-sensitive SBS
3.6 Current issues in massively parallel sequencing
4 Massively parallel sequencing – single-molecule sequencing
4.1 Reversible single-dye terminator SBS
4.2 Zero-mode waveguide sequencing with immobilized polymerase
4.3 Nanopore sequencing
5 Future perspectives
References