
دانلود رایگان مقاله DNA قدیمی و تاریخچه انسان
ما مطالعه اطلاعات ژنومی را که توسط توالی یابی فسیل های انسانی فراهم شده است، مرور میکنیم با تاکید بر اطلاعات یگانه ای که DNA قدیمی aDNA میتواند درباره ی تاریخچه ی جمعیت انسان و نزدیکترین خویشاوندان آن فراهم کند. ما بر توالی های ژنوم هسته ای که در سال های اخیر چاپ شده است تمرکز کردیم. در بسیاری از موارد، بخصوص در قطب شمال و آمریکای شمالی و جنوبی و اروپا ، aDNA الگوی جمعیتی تاریخی به این صورت نشان داد که توسط آنالیز ژنوم امروزه به تنهایی فاش نمیشود. DNA قدیمی از hominins (شبه انسان های) قطب شمال ، تاریخچه ی غنی از اختلاط بین انسان های مدرن امروزه ، Neanderthals (انسان های غار نشین) و Denisovans را نشان داد و به ما اجازه داد که فرآیند انتخابی پیچیده را رها کنیم. اطلاعات از مطالعات aDNA، در حال حاضر نزدیک اشباع شدن است و ما معتقدیم که توالی یابی aDNA آینده به تغییر درک ما از تاریخچه ی hominins ادامه میدهد.
تکامل ژنومیکس به خوبی در حال انجام است. در زمانی که اولین توالی های ژنوم انسان فراهم شد، تقریبا غیر ممکن است که با 15 هزار ژنوم از مردم در سراسر دنیا با پوشش بالا توالی یابی شود، احتمالا کمی ممکن تر است که توالی های بخشی یا کل ژنوم از صدها فسیل انسانی مدرن ، چندین فسیل Neanderthals ، و حتی فسیل از گروه خواهری قبلی Neanderthals که Denisovans خوانده میشوند، فراهم شود، برخی از این ژنوم های قدیمی چنان با عمق زیادی توالی یابی شده اند که خطاهای آنها ، توالی های با پوشش بالا از انسان های امروزی را ارزیابی میکند.
غنای اطلاعات ژنومی امروزه و قدیمی به خوبی آنچه تقاضای ژنتیک¬دانان جمعیت است را افزایش داده است. وقتی که تعداد کمی لوکوس با استفاده از لوکوس مارکر مطالعه شد، به طور عمده گروه های خونی، آلوزیم ها و آمار توصیفی ناخالص - میکروساتلایت ها از قبیل هتروزیگوتی، Wright’s FST ، فاصله های ژنتیکی متنوع مطالعه شدند که برای شناسایی الگوهای گسترده از تمایز جمعیت کافی بودند. این روشهای کلاسیک توسط Luca Cavalli-Sforza و بسیاری از همکارانش برای اولین بار استفاده شدند. در 1964 Cavalli-Sforza و همکارن درخت فیلوژنتیک از 15 جمعیت های انسان بر مبنای 20 آلل در 5 لوکوس اکثرا گروه های خونی، چاپ کردند که برای آنها اطلاعات چاپ شده ی کافی وجود داشت. مولفان درخت را بر نقشه ی دنیا اضافه کردند که مسیر پراکنش گذشته را پیشنهاد کنند. نقشه ی آنها شامل بیشتر مطالعات اخیر بر مبنای بیشتر اطلاعات بود. تنها ارتباط Maori به آمریکایی های بومی ، با نظریه ی رایج پذیرفته شده مغایر بود که Maori از جزیره ی Polynesians آمده است.
در حال حاضر، نه تنها می توان الگوهای گسترده روابط بین جمعیت ها را روشن کننده بلکه آنها میتوانند پاسخهای مفصل به سوالات تاریخی مرتبط با دیرینه شناسی را فراهم کنند. از چه زمانی ، کجا و چه منبعی ، جمعیت های انسانی مخصوص بوجود آمدند؟ چه کسی با انها آمیزش کرد و چه زمانی آمیزش رخ داد؟ تغییرات واضح در دیرینه شناسی ، نتایج جایگزینی جمعیت یا ابداع فرهنگی را ثبت میکنند؟ آیا فرهنگ های گذشته فرزاندان ژنتیکی از خود به جا گذاشته اند. همانطور که ما بحث خواهیم کرد، آنالیز DNA قدیمی در پاسخ چند تا از این سوالات موفق بوده است اما همچنین سوالات جدیدی در فرآیند را به وجود می آورد. aDNA یک بعد موقتی از مطالعات ژنتیکی را فراهم میکند که به تنهایی توسط ژنوم امروزه قابل دسترسی نیست و تنها می داند که همه علائم aDNA کشف شده است.
آلودگی
یکی از مشکلات عمده که توالی یابی aDNA hominin را برای چندین سال جلوگیری کرد، آلودگی بود. مواد ژنتیکی استخراج و توالی یابی بافت نمونه زنده به طور انفرادی شامل طیف وسیعی از قطعات DNA آن فرد است اگر فعالیت های استاندارد آزمایشگاهی پیروی شود. برخلاف آن، به این دلیل که aDNA بسیار نادر و قطعه قطعه است، بیشتر مواد ژنتیکی که از فسیل ها گرفته میشوند تمایل دارند که اگزوژنز باشند معمولا از میکروب های محیطی یا انسان هستند که فسیل ها را به کار گرفتند. نوع بعدی DNA مخصوصا دردسرساز است زیرا DNA انسان امروزه در توالی مشابه aDNA اندوژنز از فسیل های hominin است و میتواند انحراف در آنالیز پایین دست را معرفی کند.
اگرچه برخی از مطالعات اولیه aDNA هسته ای از hominin دیرینه با آلودگی مشکل داشت، اختراع های محاسباتی و تجربی برای کاهش دادن اثر در مطالعات معاصر انجام شدند. در دهه ی گذشته، محققان دو مجموعه ی گسترده از روش ها را برای مطالعه ی توالی های مفید گذشته ایجاد کردند.
اول، تمرین استاندارد برای استخراج aDNA تحت شرایط اتاق استریل شامل اشعه ی uv ، تیمار سفید کننده سطح و سیستم تصفیه ی هوا بود بنابراین آمادگی DNA اگزوژنز در عصاره ی فسیل کم کردیم. به علاوه، در زمان ساخت کتابخانه ی DNA، دانشمندان، آداپتورهای یگانه ساختار کتابخانه ی DNA برای نشاندار کردن مولکول ها ترکیب کردند که در زمان استخراج حاضر است، برای جلوگیری از مولکول های اضافه به طور تصادفی در طول گام های توالی یابی اضافه میشود که با مولکول های اندوژنز اشتباه میشوند.
ثانیا، بعد از اینکه DNA توالی یابی شد، چندین ابزار بیوانفورماتیک می تواند استفاده شود تا یا قطعات توالی را حذف کند یا آمادگی این قطعات حاضر در کتابخانه ی DNA تخمین بزند. کار رایج این است که نرخ آلودگی را با استفاده از DNAمیتوکنرایی تخمین بزنند که خیلی از DNA هسته ای فراوان تر است و از این رو توالی ان با پوشش خیلی بالاتراز DNA هسته ای انجام میشود. برای جمعیت هایی که خیلی مشتق شده اند (Neanderthals) می توان موقعیت های تشخیصی استفاده کرد که دو گروه را مشخص میکند و ارزیابی میکند که چه تعداد قطعات توالی ناسازگار در هر موقعیت وجود دارد. برای جمعیت های مدرن انسانی (اروپایی های قدیمی) میتوان قطعات توالی را چک کرد که از توالی اجماعی انشعاب پیدا کردند یا آنهایی که علائم مولکولی پایدار با aDNA دارند. همچنین روش های تخمین نرخ آلودگی تصادفی بیشتری وجود دارند که اطلاعات بزرگتری شامل کروموزوم های جنسی و کل ژنوم اتوزوم را استفاده میکنند. به علاوه، میتوان الگوهای دآمیناسیون در انتهای قطعات - مرگ شیمیایی aDNA- برای اینکه قطعاتی را که این علائم را نشان نمی دهند فیلتر کند که احتمالا قدیمی نیستند.
Hominins دیرینه
توالی یابی و آنالیز ژنوم از Neanderthals و وابستگان آن نقص در تکامل را نشان داد. ابتدا، سوال از زادآوری بین Neanderthals و انسان مدرن بود –که با 30 y دیرینگی از هم جدا میشوند، اکنون به طور قانع کننده ای پاسخ داده شده است. به علاوه، یک گروه خواهری Neanderthals که Denisovans نامیده میشوند ، کشف شدند و رابطه ی آنها با Neanderthals و انسان تثبیت شد.
Neanderthals . علیرغم اینکه محدوده ی Neanderthals و انسان مدرن در اروپا و آسیای غربی برای حداقل 10000 y همپوشانی داشته، هیچ مدرک قدیمی پذیرفته شده وجود ندارد که Neanderthals و انسان مدرن تعامل یا زاد و ولد کردند. Krings و همکاران دریافتند که توالی یابی DNA میتوکندریایی از فسیل Neanderthals که در بیرون از کلاد توالی های DNA میتوکندریایی قرار گرفته فراهم شده است که متشکل از همه توالی های میتوکندری از انسان های مدرن است. این الگوی دوجانبه تک نیایی در بسیاری از مطالعات دیرتر DNA میتوکندریای Neanderthals ثابت شده است.اگرچه، درخت DNA میتوکندریایی با این فرضیه پایدار است که آمیزشی بین دو گروه وجود ندارد، اما این موضوع مدارک واضح کافی بر علیه آن فراهم نمیکند. در حقیقت، تک نیایی دو جانبه با احتمال بالایی دیده شده است حتی اگر آمیزش وجود داشته باشد. قبل از توالی یابی ژنوم اولیه Neanderthals ، آنالیز نمونه های انسانی امروزه نشان داده است که احتمالا سطوح بالای قدمت در هر دو ژنوم آفریقای غربی و اروپا وجود دارد که احتمالا از گروه hominin مشتق شده است.
We review studies of genomic data obtained by sequencing hominin fossils with particular emphasis on the unique information that ancient DNA (aDNA) can provide about the demographic history of humans and our closest relatives. We concentrate on nuclear genomic sequences that have been published in the past few years. In many cases, particularly in the Arctic, the Americas, and Europe, aDNA has revealed historical demographic patterns in a way that could not be resolved by analyzing present-day genomes alone. Ancient DNA from archaic hominins has revealed a rich history of admixture between early modern humans, Neanderthals, and Denisovans, and has allowed us to disentangle complex selective processes. Information from aDNA studies is nowhere near saturation, and we believe that future aDNA sequences will continue to change our understanding of hominin history.
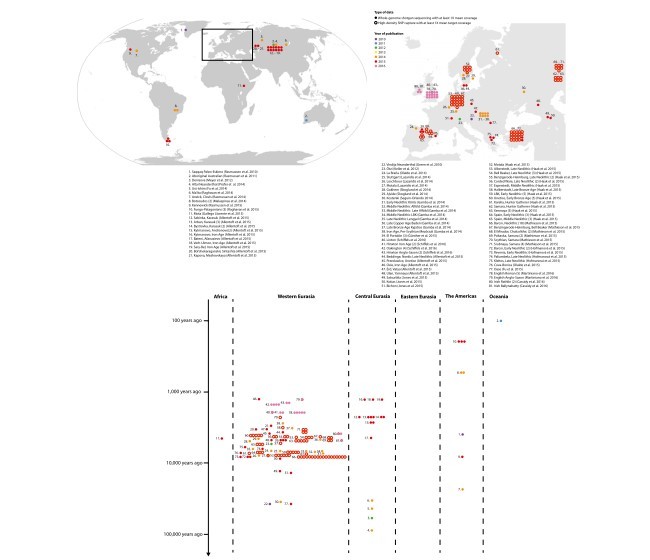
The genomics revolution is well under way. At the time that the first human genomic sequences were obtained (1, 2), it was almost inconceivable that within 15 y thousands of genomes from people around the world would be sequenced, many to a high depth of coverage (3). It was probably even less conceivable that partial or complete genomic sequences would be obtained from hundreds of modern human fossils (4–6), several Neanderthal fossils (7, 8), and even fossils of a previously unknown sister group of Neanderthals, called Denisovans (9, 10) (Fig. 1). Some of these ancient genomes have been sequenced to such high depth that their error rates rival those of high-coverage sequences from present-day humans.
The wealth of present-day and ancient genomic data has greatly increased what is demanded of population geneticists. When relatively few loci could be studied using marker loci—chiefly blood groups, allozymes, and microsatellites—gross descriptive statistics, such as heterozygosity, Wright’s FST, and various genetic distances were sufficient to characterize broad patterns of population differentiation. Application of these classic methods was pioneered by Luca Cavalli-Sforza and his many collaborators. As early as 1964, Cavalli-Sforza et al. (11) published a phylogenetic tree of 15 human populations based on a total of 20 alleles at 5 loci, mostly blood groups, for which adequate published data were available. The authors superimposed the tree on a world map to suggest past dispersal routes. Their map is surprisingly consistent with more recent studies based on vastly more data. Only the connection of Maori to Native Americans disagrees with currently accepted theory, that the Maori descended from Polynesians (12).
At present, not only can geneticists elucidate broad patterns of relationship among populations, but they can also provide detailed answers to historical questions of relevance to archeology and paleoanthropology. When, where, and from what source did particular human populations arise? Who admixed with whom and when did the admixture take place? Are obvious changes in the archaeological record the result of population replacement or cultural innovation? Did past cultures leave any genetic descendants? As we will discuss, analysis of ancient DNA (aDNA) has been successful in answering several of these questions, but has also raised new questions in the process. Importantly, aDNA provides a temporal dimension to genetic studies that would be inaccessible with present-day genomes alone, and only now is the full significance of aDNA being explored.
Contamination
One of the major problems that prevented the widespread sequencing of hominin aDNA for several years was contamination. Genetic material extracted and sequenced from a tissue sample of a living individual will consist largely of DNA fragments from that individual (i.e., endogenous fragments) if standard laboratory practices are followed. In contrast, because aDNA is so scarce and fragmented, most of the genetic material extracted from fossils tends to be exogenous, usually either from environmental microbes or humans who handled the fossil (13). The latter type of DNA is especially troublesome, as presentday human DNA is similar in sequence to endogenous aDNA from hominin fossils, and can introduce biases in downstream analyses.
Although some of the first studies of nuclear aDNA from archaic hominins had problems with contamination (14, 15), there have been substantial experimental and computational innovations for mitigating its effect in contemporary studies. In the past decade, researchers have developed two broad sets of approaches to correct for contamination in their aDNA samples, allowing for the study of previously unusable sequences.
First, it is now a standard practice to extract aDNA under strict clean-room conditions—including UV radiation, bleach treatment of surfaces, and filtered air systems—so as to minimize the proportion of exogenous DNA in the fossil extracts (13). Additionally, at the time of DNA library construction, scientists incorporate unique adapters to tag molecules that are present at the moment of extraction (16), to prevent additional molecules accidentally added during subsequent sequencing steps from being confused with endogenous molecules.
Second, after the DNA has been sequenced, several bioinformatic tools can be used to either remove contaminant reads or estimate the proportion of those reads present in a DNA library. A common practice is to estimate the rate of contamination using mitochondrial DNA (mtDNA), which is much more abundant than nuclear DNA and hence is sequenced to a much higher coverage than nuclear DNA. For highly divergent populations (e.g., Neanderthals), one can use diagnostic positions that distinguish the two groups and assess how many discordant reads are present at each position (17, 18). For modern human populations (e.g., ancient Europeans), one can check for reads that diverge from the consensus sequence or that do not contain molecular signatures consistent with aDNA (19, 20). There are also more sophisticated contamination rate estimation methods that use larger subsets of the data, including sex chromosomes (21, 22) and entire autosomal genomes (7, 23). Additionally, one can use patterns of cytosine deamination at the ends of fragments—a postmortem chemical damage typical of aDNA—to filter out sequenced reads that do not display this signature and are therefore not likely to be ancient (24).
Archaic Hominins
The sequencing and analysis of genomes from Neanderthals and their relatives has been nothing short of revolutionary. First, the question of interbreeding between Neanderthals and modern humans—posed by paleoanthropologists over 30 y ago—has now been convincingly answered (7, 8). Additionally, a sister group of Neanderthals, called Denisovans, was discovered and its relationship to Neanderthals and humans established (9, 10).
Neanderthals. Despite the overlapping ranges of Neanderthals and modern humans in Europe and western Asia for at least 10,000 y, there was no widely accepted archaeological evidence that Neanderthals and modern humans interacted or interbred. Krings et al. (25) found that mtDNA sequences obtained from a Neanderthal fossil lay outside the clade composed of all mtDNA sequences from modern humans. This pattern of reciprocal monyphyly has been confirmed in many later studies of Neanderthal mtDNA (17). Although the mtDNA tree was consistent with the hypothesis that there was no admixture between the two groups, it did not provide conclusive evidence against it. In fact, reciprocal monophyly would be seen with significant probability even if there had been substantial admixture (26). Before the sequencing of the first Neanderthal genome, an analysis of presentday human samples had indicated there might have been high levels of archaic ancestry in both European and West African genomes, likely stemming from a diverged hominin group (27).
آلودگی
Hominins دیرینه
aDNA انسان های مدرن
بحث
منابع
Contamination
Archaic Hominins
aDNA from Modern Humans
Discussion
References