
دانلود رایگان مقاله ابزاری از MATLAB برای غنی سازی مسیر با استفاده از امتیاز نظم مسیر
چکیده
پیشینه: رسیدگی به مقدار وسیع دادههای تظاهر ژنی تولیدشده توسط روشهای نمای نسخهبرداری ژنوم، یک کار چالشبرانگیز است که نیازمند ترکیبی آگاه از روشهای پیشپردازش، فیلتراسیون و تحلیل دارد اگر که قرار باشد نتایج زیستشناختی معناداری حاصل شود. برای مثال، طیفی از آمار سنتی و رویکردهای تحلیل مسیر محاسبهای برای شناسایی فرایندهای بسیار ارائهشده در دادههای ریزآرایه بهدستآمده از حالتهای مختلف بیماری استفادهشده است. اگرچه، اکثر این رویکردها تمایلی در بهرهبرداری از کل طیف داده تظاهر ژنی یا روابط مختلف و وابستگیها ندارند. قبلاً، ما ابزار تحلیل غنیسازی مسیر را در MATLAB که یک امتیاز نظم مسیر (PRS) را با در نظر گرفتن علامتدهی توپولوژی مسیر و بیش نمایندگی و بزرگی ژنهایی که بهطور متفاوتی ظاهرشدهاند به دست میدهد. در اینجا، این رویکرد را برای شامل شدن مسیر متابولیک گسترش دادیم و استفاده از رابط کاربر گرافیکی (GUI) را توصیف کردیم.
نتایج: با استفاده از تعدادی از جایگاههای ریزآرایه و گونهها، مصرفکنندگان قادرند تا امتیازات PRS را به همراه امتیاز z مطابق برای مقایسه محاسبه کنند. ارزیابی بیشتر اهمیت مسیر ممکن است برای افزایش اعتماد به مسیرهای بهدستآمده انجام شود و مصرفکنندگان میتوانند دایره المعارف Kyoto نمودارهای مسیر ژن و ژنوم را که برای تأکید بر ژنهای نهفته علامتگذاری شده است، ببینند.
نتیجهگیری: ابزار PRS، فیلتری برای منزویسازی بینشهای زیستی از دادههای نسخهبرداریشده پیچیده ارائه میکند.
پیشینه
بهطور فزایندهای، روشهای نمای نسخهبرداری با بازده بالا (ریزآرایهها یا بهطور فزایندهای، دنبالههای RNA) تحقیقات علوم حیاتی مدرن را شکل میدهد. چنین روشهایی، یک دوربین مولکولی فراهم میکند که تصاویری از سراسر ژنوم فعالیت ژنتیکی میگیرد. اگرچه، دادههای تحلیل مؤثر ریزآرایهها، چالشهایی را مخصوصاً در رسیدگی به تعداد زیادی از ژنهایی که بهطور همزمان مطالعه میشوند ارائه میدهد.
تحلیل تظاهر ژن درزمینهٔ دانش برگزیده یا "تحلیل مسیر ناشی از پایه دانش" ازآنجاییکه این مسئله باعث کاهش در فضای تحقیقاتی از هزارها ژن تا زیرمجموعهای از فرایندهای زیستشناختی که برای تفسیر انسانی بیشتر مهار شدنیتر است ضروری است [1]. طبق Khatri و همکارانش [2]، رویکردهای غنیسازی مسیر میتواند به سه نسل تقسیمبندی شود:
1- تحلیل بیش نمایندگی (ORA): این تحلیل یک مسیر را با در نظر گرفتن نسبت ژنهایی که بهصورت متفاوت ظاهرشده (DEG) و در هر مسیر مربوط به نسبت تمام ریزآرایههای DEG مشاهده میشود، ثبت میکند. این تحلیل برای ابزارهای تحلیل مسیر متعددی منجمله GenMAPP [3], GoMiner [4], [5] Onto-Express and FatiGo [6] به کار میرود.
2- امتیازدهی طبقه عملکردی (FCS): FCS یک امتیاز را به هر ژن بر اساس تظاهر آن، در مسیر میدهد که با آن امتیاز مسیر بر اساس امتیازات تمام ژنها در مسیر محاسبه میشود. تعدادی از روشهای FCS از طریق ابزارهای مستقل مانند GSEA [7]، SigPathway [8] و SAFE [9] یا ابزارهای وب مانند T-profiler [10]، Gazer [11] و GeneTrail [12] اجرا میشود.
3-رویکردهای مبتنی بر توپولوژی مسیر (PT): این رویکردها از توپولوژی مسیرها با دادن اوزان برای اتصالات از پیش تعیینشده بین ژنها که امتیازدهی مسیر را تشکیل میدهند، بهره میگیرند. برخی رویکردهای مبتنی بر توپولوژی در دهههای گذشته در ادبیات توصیفشده است. طبق Mitrea et al [13]، رویکردهای مبتنی بر PT، در روشی که اطلاعات توپولوژی مسیر را به امتیاز مسیر ترجمه میکند متفاوتاند. برخی روشها تنها از دادههای توپولوژی ژنهایی که بهصورت متفاوت ظاهرشدهاند (DEG) در امتیاز غنیسازی استفاده میکنند (مثلاً، MetaCore[14]، EnrichNet [15])، درحالیکه دیگر روشها (منجمله، SPIA [16] و GANPA [17]) از دادههای تظاهر DEG به همراه دادههای توپولوژی استفاده میکنند. متناوباً، برخی روشها از دادهای تظاهر ناشی از تمام ژنهای ریزآرایه استفاده میکنند چه بین شرایط تغییر بکنند یا نکنند، برای مثال، PathOlogist [18]، DEGraph [19] و ACST [20]. نکته مهم اینکه برخی ابزار مبتنی بر PT تنها از توصیفهای علامتدهی مسیر مانند Pathway-Express [21]، NetGSA [22]، ScorePAGE [23]، TAPPA [24]، MetPA [25] و Clipper [26] استفاده میکنند.
پیشازاین، یک روش غنیسازی مسیر جدید را ارائه کردیم که در آنهم توپولوژی مسر و هم بزرگی تظاهر ژن، ایجاد امتیاز نظم مسیر (PRS) را تغییر میدهد [27]. مخصوصاً، با ترکیب دادههای تغییر کل برای آن نسخههایی که از آستانه اهمیت تجاوز میکنند و با در نظر گرفتن پتانسیل تظاهر ژن تغییریافته در اثرگذاری بر نسخهبرداریهای پاییندست، مسیرهایی را شناسایی کردیم که به فرایند پاتوفیزیولوژیک تحت بررسی مرتبط است. رویکرد ما تعدادی از مسائل را که بهطور بالقوه روشهای غنیسازی را تضعیف میکند در نظر میگیرد. ما گامهایی برای کاهش تأثیر خطاها در نگاشت شناسه و کاهش خطای ایجادشده توسط مسیرهای اضافی (مانند نمونههای چندگانه یک ژن) برداشتیم. روشهای توپولوژی نیز باید بهصورت مؤثر به حلقهها رسیدگی کنند، لذا ما از الگوریتم جستجویی که از نظریه گراف ناشی شده است برای حل این مشکل استفاده کردیم. همچنین احساس کردیم که تقسیمبندی دلخواهانه فرایندها به نظم بالا یا پایین، ازآنجاییکه تغییرات در تظاهر ژن احتمالاً در سراسر مسیرها توزیع میشود، ساختگی است و ازاینرو ارزیابی ما، یک ارزیابی کلی اثر بود.
در اینجا، اجرای رویکرد PRS خود را بهعنوان یک ابزار مستقل که به مصرفکننده نهایی گزینه واردکردن داده را از جایگاهها و انواع مختلف ریزآرایه میدهد، توصیف کردیم. این ابزار هم امتیازات z و هم PRS را به دست میدهد، تحلیل آماری ارائه میکند و اجازه مرور راههایی که دارای ژنهای نهفتهای هستند که بارنگهای مختلف علامتگذاری شدهاند میدهد. گزارش خود را با این مطلب که مصرفکنندگان قادرند تا هم مسیرهای متابولیک و هم علامتدهی را تقویت کنند، ارتقا میدهیم.
اجرا
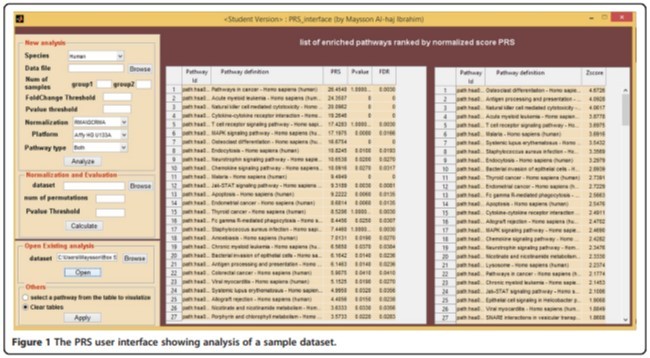
رویکرد PRS در MATLAB اجرا شد. مصرفکنندگانی که به محیط MATLAB دسترسی ندارند، میتوانند کامپایلر اجرای MATLAB (MRC) را دانلود کنند تا نرمافزار توصیفشده در اینجا را با یک GUI کاربرپسند بهکارگیرند. رابط PRS (شکل 1) توابع متعددی را برای مصرفکنندگان ارائه میدهد:
پیشپردازش دادههای ریزآرایه
ما یک فیلتر را برای نرمالسازی دادهها از جایگاهها مختلف مجدداً مهندسی نکردیم، در عوض، مصرفکنندگان ابتدا باید دادههای نسخهبرداریشده را با استفاده از یکی از ابزارهای بیشمار موجود پیشپردازش کنند. دادهها باید در قالب یک صفحه گسترده (Spreadsheet) Excel باشد که در آن اولین ستون باید شناسه محقق باشد و در ستونهای بعدی باید ارزشهای تکرار شده نرمالیزه شده تظاهر را از شرایط تست و کنترل قرار داد. اطلاعات مازاد در خصوص گونهها، تعداد نمونه، تغییر کل، آستانههای تست t، روش نرمالسازی و جایگاه موردنیاز است.
Abstract
Background: Handling the vast amount of gene expression data generated by genome-wide transcriptional profiling techniques is a challenging task, demanding an informed combination of pre-processing, filtering and analysis methods if meaningful biological conclusions are to be drawn. For example, a range of traditional statistical and computational pathway analysis approaches have been used to identify over-represented processes in microarray data derived from various disease states. However, most of these approaches tend not to exploit the full spectrum of gene expression data, or the various relationships and dependencies. Previously, we described a pathway enrichment analysis tool created in MATLAB that yields a Pathway Regulation Score (PRS) by considering signalling pathway topology, and the overrepresentation and magnitude of differentially-expressed genes (J Comput Biol 19:563–573, 2012). Herein, we extended this approach to include metabolic pathways, and described the use of a graphical user interface (GUI).
Results: Using input from a variety of microarray platforms and species, users are able to calculate PRS scores, along with a corresponding z-score for comparison. Further pathway significance assessment may be performed to increase confidence in the pathways obtained, and users can view Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway diagrams marked-up to highlight impacted genes.
Conclusions: The PRS tool provides a filter in the isolation of biologically-relevant insights from complex transcriptomic data.
Background
Increasingly, high-throughput transcriptional profiling techniques (microarrays or, increasingly, RNAseq) inform modern life-science research. Such techniques provide a molecular “camera” taking genome-wide “snapshots” of genetic activity. However, the effective analysis of microarray data presents a number of challenges, in particular handling the large number of genes that are studied simultaneously.
Analysing gene expression in the context of curated knowledge, or “knowledge base-driven pathway analysis”, is critical as this guides the reduction in search space from many thousands of genes to an subset of biological processes, which are much more tractable to human interpretation [1]. According to Khatri et al [2], pathway enrichment approaches can be divided into three generations:
i. Over-representation Analysis (ORA): This scores a pathway by considering the proportion of differentiallyexpressed genes (DEGs) observed in each pathway relative to the proportion of all microarray DEGs. This is used by several pathway analysis tools, including GenMAPP [3], GoMiner [4], Onto-Express [5] and FatiGo [6].
ii. Functional Class Scoring (FCS): FCS gives a score to each gene in a pathway based on its expression, from which a pathway-score is calculated based on the scores of all the genes in the pathway. A number of FCS methods have been implemented through standalone tools such as GSEA [7], SigPathway [8], and SAFE [9], or web tools such as T-profiler [10], Gazer [11] and GeneTrail [12].
iii. Pathway Topology (PT)-based approaches: These approaches exploit the topology of pathways by giving weights to pre-defined connections between genes, which inform pathway scoring. Several topology-based approaches have been described in the literature over the past few years. According to Mitrea et al [13], PT-based approaches differ in the way they translate pathway topology information into a pathway score. Some methods use only the topology data of differentially-expressed genes (DEGs) in the enrichment score (for example MetaCore [14] and EnrichNet [15]), whereas others (including SPIA [16] and GANPA [17]) use expression data of DEGs along with the topology data. Alternatively, some methods use expression data derived from all microarray genes, whether they change between conditions or not, for example PathOlogist [18], DEGraph [19], and ACST [20]. Importantly, some PT-based tools use only signalling pathway descriptions, such as Pathway-Express [21], NetGSA [22], ScorePAGE [23], TAPPA [24] MetPA [25], and Clipper [26].
Previously, we proposed a new pathway enrichment method, in which both pathway topology and the magnitude of gene expression changes informed the creation of a Pathway Regulation Score (PRS) [27]. Specifically, by combining fold-change data for those transcripts exceeding a significance threshold, and by taking into account the potential of altered gene expression to impact upon downstream transcription, we identified those pathways most relevant to the pathophysiological process under investigation. Our approach addressed a number of issues that potentially compromise enrichment methods. We took steps to mitigate the influence of errors in ID mapping, and to reduce the bias introduced by highlyredundant pathways (i.e. multiple instances of the same gene). Topology methods also have to handle loops effectively, so we used a search algorithm derived from graph theory to resolve this problem. We also felt that arbitrarily dividing processes into either up- or downregulated was artificial as changes in gene expression are likely to be distributed throughout pathways, thus ours was an overall impact assessment.
Herein, we described the implementation of our PRS approach as a standalone tool that provides end users with the option of importing data from different microarray platforms and species. The tool yields both PRS and z-scores, provides statistical analysis, and allows browsing of pathways with impacted genes highlighted in different colours. An enhancement from our original report is that users are able to enrich both signalling and metabolic pathways.
Implementation
The PRS approach was implemented in MATLAB. Users without access to the MATLAB environment can download the MATLAB Runtime Compiler (MRC) in order to deploy the software described herein, via a user-friendly GUI. The PRS interface (Figure 1) provides users with several functions:
Preprocessing microarray data
We did not re-engineer a filter to normalise data from a variety of platforms, rather users must first preprocess transcriptomic data using one of the myriad existing tools.
چکیده
پیشینه
اجرا
پیشپردازش دادههای ریزآرایه
نمایش مسیر
امتیازدهی مسیر
ارزیابی اهمیت مسیر
تجسم مسیرهای غنیشده
UML برای مدلسازی و توصیف نرمافزار
نتایج و بحث
نتیجهگیری
منابع
Abstract
Background
Implementation
Preprocessing microarray data
Pathway representation
Pathway scoring
Pathway significance assessment
Visualizing enriched pathways
UML for modelling and software description
Results and discussion
Conclusions
References