
دانلود رایگان مقاله التهاب تورونی در بیماری پارکینسون و توانایی بالقوه ی آن به عنوان یک هدف دارویی
مطالعات انجام شده با توموگرافی تابش پوزیترون (PET) نشان دهنده ی فعال سازی میکروگلیا در نواحی مختلف مغز بیمار مبتلا به پارکینسون است. علاوه بر این، فعال سازی میکروگلیا در SNpc و جسم مخطط در انواع مختلفی از مدل های حیوانی بیماری پارکینسون مشاهده شده است. بررسی های بیوشیمیایی بیشتر سطوح بالاتری از واسطه های پیش التهابی مانند فاکتور نکروز تومور – الفا (TNF-α)، اینترلوکین 1-بتا (IL-1β) و اینترفرون گاما (IFN-γ) را در قسمت میانی مغز بیمار مبتلا به پارکینسون نشان می دهد. این داده ها با قوت نشان دهنده ی نقش اجزاء سیستم ایمنی در پاتوژنز بیماری پارکینسون است.
در شرایط فیزیولوژیکی، وضعیت غیرفعال میکروگلیا از طریق انواع مختلفی ازحدواسط های ایمنی نظیر CX3CL1، CD200، CD22، CD47، CD95 و مولکول چسبندگی سلولی عصبی (NCAM) حفظ می شود که عمدتا توسط سلول های عصبی تولید می شوند. جالب است که، گیرنده های این مولکول ها تقریبا به طور انحصاری توسط میکروگلیا در CNS بیان می شوند، که نشان دهنده نقش حیاتی میانکنش های نورون- میکروگلیا در تنظیم التتهاب نورون است. علاوه بر این، سیگنال CX3CL1-CX3CR1 تنظیم کننده ی منفی فعال سازی میکروگلیاست و نورون های DA را در برابر تخریب ناشی از نوروتوکسین ها مخافظت می کند. نقص در CX3CL1 یا CX3CR1 در شرایط in vivo باعث افزایش نوروتوکسیسیتی ناشی از تیمار سیتمیک لیپوپلی ساکارید (LPS) می شود و مرگ سلولی نورون های DA را در SNpc حیوانات مدل بیماری پارکینسون افزایش می دهد. همچنین از کار افتادن سیگنالینگ CD200-CD200R نیز فعال شدن میکروگلیا را افزایش داده و تخریب نورون های DA را در حیوانات مدل پارکینسون تشدید می کند.
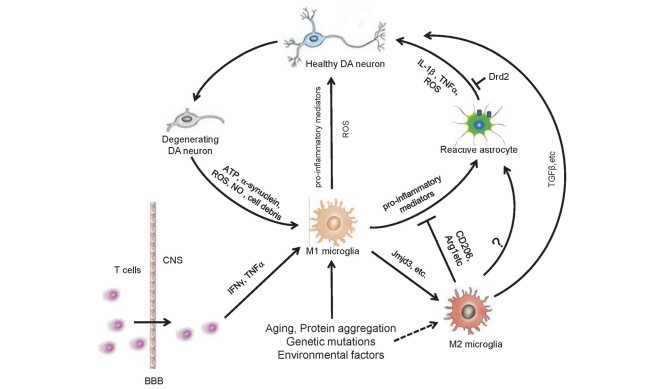
به نظر می رسد که میکروگلیای فعال شده حداقل در مراحل اولیه ی فرایند تخریب سلول های عصبی، می تواند برای میزبان مفید باشد. علاوه بر این، نشان داده شده است که سرکوب Jmjd3، که برای پولاریزاسیون میکروگلیای M2 ضروری است، در جسم سیاه (SN) در شرایط in vivo باعث فعال شدن بیش از حد میکروگلیا و تشدید تولید دوپامین و مرگ نورون ها در حیوانات مدل بیماری پارکینسون می شود، که نشان دهنده ی نقش حافظتی میکروگلیای M2 در این فرایند است. با این وجود، فعالیت بیش از حد میکروگلیا به مدت طولانی در مغز بیماران مبتلا به پارکینسون به میزان زیادی بیان گروه بزرگی از سیتوکین های پیش التهابی مانند TNF-α، IL-1β، اینترلوکین-6 (IL-6) و IFN-γ را افزایش می دهد که در تخریب نورون های نیگرال DA نقش دارد. با پیشرفت بیماری، مولکول هایی نظیر α سینوکلین، ATP و متالوپروتئیناز3 (MMP 3) که از نورون های دژنرژیک DA آزاد می شوند، باعث تقویت فعال سازی میکروگلیا، تقویت پاسخ های عصبی –التهابی در مغز و موجب تشدید فرایندهای نورودژنراسیون می گردند و به تشکیل یک چرخه ی نادرستی از نورودژنراسیون می شود. میکروگلیای فعال شده در اطراف ترکیبات حاوی α سینوکلین در بسیاری از مناطق مغز بیماران مبتلا به پارکینسون ابناشته می شود. این سلول ها به احتمال زیاد بوسیله ی α سینوکلین های بیش از حد، جهش یافته یا بد تاخورده فعال می شوند که منجر به افزایش تولید و انتشار سیتوکین های التهابی می شود. بنابراین، نوروتوکسیسیتی القاء شده توسط α سینوکلین های مازاد یا بدتاخورده ممکن است بوسیله ی پاسخ های التهابی میکروگلیا باشد.
ATP، به عنوان یک انتقال دهنده ی عصبی پورینرژیک، نیز قادر است انواع مختلفی از عملکردهای میکروگلیا را وساطت کند. مهاجرت میکروگلیا به مناطق آسیب دیده و التهابی توسط ATP آزاد شده از نورون های آسیب دیده و آستروسیت های همسایه کنترل می شود. علاوه بر این، ATP به گیرنده ی P2Y متصل می شود که عمدتا توسط میکروگلیا بیان می شود و تولید سطوح بالایی از IL-1β، TNF-α و نیتریک اکسید (NO) را تحریک می-کند. پروتئین دیگر تولید شده توسط نورون های دژنراتیو MMP 3 است که نقش مهمی در تنظیم وضعیت فعال میکروگلیا، حداقل در شرایط آزمایشگاهی، دارد. بیان بیش از حد MMP 3 در کشت های ترکیبی میکروگلیا - نورون موجب فعال شدن میکروگلیا و افزایش واکنش استرس اکسیداتیو می گردد. این در حالی است که موش های بدون MMP-3 که N-methyl-4-phenyl-1,2,3,6-tetrahydro- pyridine (MPTP) به آن تزریق شده بود سطح پایینی از دژنراسیون نورونی نیگراستیراتیال، فعال شدن میکروگلیا و تولید سوپراکسید را نشان دادند. این داده ها این مطلب را که میکروگلیا مهم ترین بازیگر در التهاب نورونی در پاتوژنز بیماری پارکینسون است و MMP-3 نقش مهمی در دژنراسیون نورونی دوپامینرژیک دارد را تایید می کنند.
فنونیپ های فعال شدن میکروگلیا در بیماری پارکینسون
شواهد نشان می دهد که میکروگلیای فعال شده دارای دو فنوتیپ متفاوت است که عبارتند از فنوتیپ M1 ( پیش التهابی) و فنوتیپ M2 (ضدالتهابی). این وضعیت های متفاوت میکروگلیای فعال با ترشح دسته های مختلفی از سیتوکین ها مشخص می شوند. نشان داده شده است که تیمار با LPS / IFN γ موجب فعال شدن M1 می شود، در حالی که تیمار با IL 4 / IL 13 موجب فعال شدن M2 در میکروگلیا می گردد. فعال سازی کلاسیک (M1) میکروگلیا با تولید سیتوکین های پیش التهابی مانند TNF-α، IL-1β، IL-6، IL-12 و سایر مولکول های سایتوتوکسیک مانند سوپراکسید، NO و گونه های فعال اکسیژن (ROS)، مشخص می شود که در ازدیاد پاسخ های پیش التهابی در طول صدمات و عفونت ها نقش دارند. این در حالی است که میکروگلیای M2 از طریق مقابله با میکروگلیای M1 و تحریک بازسازی بافت، نقش سرکوب کننده ی ایمنی را بازی می کند. میکروگلیای M2 انواعی از سیتوکین های دارای خاصیت التهابی مانند IL-4، IL-13، IL-10 و TGF-β را تولید می کند. اشکال مختلف میگروگلیا از روی الگوی بیان ژن آنها نیز قابل تشخیص است. برای مثال، Arg1، FIZZ1 (که با عنوان RELM-α نیز شناخته می شود) و CD206، در فاز M2 میکروگلیا بیان می شوند. بیان Arg1، FIZZ1 و Chi3l3 احتمالا بوسیله ی سیتوکین ها تنظیم می شود ، زیرا سطوح آنها به طور قابل توجهی در میکروگلیای کشت اولیه یا مناطق قشر مغزی و پیشانی مغز موش پس از تحریک IL 4 افزایش می یابد.
چه عواملی بر فنوتیپ میکروگلیایی M1 / M2 در مورد بیماری پارکینسون تأثیر می گذارد؟ پروتئین های بد تاخورده و توکسین های محیطی باعث فعال شدن میکروگلیا به سمت فنوتیپ M1 در مدل حیوانی بیماری پارکینسون می شوند. مصرف مزمن MPTP منجر به کاهش پیشرونده بیان CD206 می شود، که نشان دهنده ی کاهش بیان فاز M2 میکروگلیا در پیشرفت بیماری پارکینسون است. برعکس، تیمار با IL 4 تا حد زیادی بیان میکروگلیایی دمتیلاز هیستون H3K27me3 demethylase (Jmjd3) را افزایش می دهد که در تنظیم تغییرات اپی ژنتیک کروموزوم ها دخیل است و در بیماری های انسانی مختلف نقش دارد. سطوح بیان ژن های مارکر M2 مانند Arg1 و CD206 پس از برداشتن Jmjd3 در سلول میکروگلیای N9 به طور قابل توجهی کاهش می یابد، که نشان دهنده نقش اساسی Jmjd3 برای قطبی شدن میکروگلیای M2 است. خاموش کردن Jmjd3 در شرایط invivo از دست رفتن نورون DA در SNpc موش های مدل تیمار شده با MPTP را با لغو فعال سازی میکروگلیای M2 تشدید می کند.
محیط CONDITIONED (CM) تهیه شده از میکروگلیا M1 فاز N9 موجب افزایش مرگ و میر نورون های DA می شود، در حالی که CM مخلوط از هر دو سلول M1 و M2 اثرت نوروتوکسیک M1-CM را تغییر می-دهد. بررسی های پیشین نشان داد که تعداد زیادی از میکروگلیای فعال شده در مراحل اولیه ی پس از صدمه ژن-های مربوط به فاز M2 را بیان می کند. ژن های امضای (شاخص) M1، به تدریج و در مراحل بعدی غالب می-شوند. این مشاهدات جالب نشان می دهد که تعادل فنوتیپ های مختلف میکروگلیای فعال شده در بیماری پارکینسون بسیار مهم هستند (شکل 1). به نظر می رسد تداخل در پیشرفت بیماری پارکینسون با دستکاری انتقال وضعیت فعال سازی میکروگلیا یک استراتژی نویدبخش باشد. با وجودی که داده های موجود نقش مؤثر M1 و M2 در پاتوژنز PD را در مدل های حیوانی نشان می دهند،اما نتایج مشابهی در بیماران مشاهده نشده است. در مقالات بعدی باید به تحقیقات انجام شده در این زمینه پرداخته شود.
Abstract
Parkinson’s disease (PD), the second most common age-associated neurodegenerative disorder, is characterized by the loss of dopaminergic (DA) neurons and the presence of α-synuclein-containing aggregates in the substantia nigra pars compacta (SNpc). Chronic neuroinflammation is one of the hallmarks of PD pathophysiology. Postmortem analyses of human PD patients and experimental animal studies indicate that activation of glial cells and increases in pro-inflammatory factor levels are common features of the PD brain. Chronic release of proinflammatory cytokines by activated astrocytes and microglia leads to the exacerbation of DA neuron degeneration in the SNpc. Besides, peripheral immune system is also implicated in the pathogenesis of PD. Infiltration and accumulation of immune cells from the periphery are detected in and around the affected brain regions of PD patients. Moreover, inflammatory processes have been suggested as promising interventional targets for PD and even other neurodegenerative diseases. A better understanding of the role of inflammation in PD will provide new insights into the pathological processes and help to establish effective therapeutic strategies. In this review, we will summarize recent progresses in the neuroimmune aspects of PD and highlight the potential therapeutic interventions targeting neuroinflammation.
Introduction
Parkinson’s disease (PD) is an age-related neurodegenerative disorder characterized clinically by resting tremor, slowness of movement, rigidity and postural instability and pathologically by the progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) [1–4]. Deposition of protein aggregates containing α-synuclein (termed Lewy bodies) is evident in multiple brain regions of advanced PD patients [5]. The etiology of PD has not yet been fully understood. Although a variety of possible pathogenetic mechanisms have been proposed over the years, including excessive release of oxygen free radicals during enzymatic dopamine breakdown, impairment of mitochondrial function, loss of trophic support, abnormal kinase activity, disruption of calcium homeostasis, dysfunction of protein degradation and neuroinflammation, the pathogenesis of PD is still largely uncertain [6–8]. However, emerging evidence indicates that sustained inflammatory responses, T cell infiltration and glial cell activation are common features of both human PD patients and animal models of PD and play vital roles in the degeneration of DA neurons [9, 10], which suggests the possibility of developing potential therapies for PD by targeting the inflammatory processes. In this review, we will focus on the role of inflammation in the progression of PD and the potential application of anti-inflammatory medications in the treatment of this devastating disorder.
Microglia-mediated inflammation in PD
Microglia is one of the major cell types which are involved in the inflammatory responses in the central nervous system (CNS) [11]. In 1988, McGeer et al. showed the presence of reactive microglia in the SNpc of human post-mortem brain tissue, which is the evidence revealed for the first time suggesting the involvement of neuroinflammation in PD pathogenesis [12]. Positron emission tomography (PET) studies also indicate that there is pronounced activation of microglia in various regions of PD brain [13, 14]. Moreover, activation of microglia in the SNpc and striatum is profound in various types of PD animal models [15–17]. Further biochemical analysis reveals higher levels of proinflammatory mediators including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interferon-gamma (IFN-γ) in the midbrain of PD patients. These data strongly suggest the involvement of immune components in PD pathogenesis.
Under physiological conditions, the quiescent state of microglia is maintained by a variety of immunomodulators, such as CX3CL1, CD200, CD22, CD47, CD95 and neural cell adhesion molecule (NCAM), which are produced mainly by neuronal cells [18–26]. Interestingly, the receptors for these molecules are almost exclusively expressed by microglia in the CNS, indicating the critical role of neuron-microglia interactions in the regulation of neuroinflamamtion [18–26]. For instance, CX3CL1-CX3CR1 signaling negatively regulates microglial activation and protects DA neurons from degeneration induced by neurotoxins [27, 28]. Deficiency of CX3CL1 or CX3CR1 in vivo results in increased neurotoxicity induced by systemic lipopolysaccharide (LPS) treatment and enhanced cell death of DA neurons in the SNpc of animal PD models [28, 29]. Likewise, dysfunction of CD200-CD200R signaling also increases the activation of microglia and exacerbates the degeneration of DA neurons in rat PD models [30, 31].
It has been proposed that activated microglia may be beneficial to the host, at least in the early phase of neurodegeneration process [12, 32–34]. For instance, it has been shown that suppression of Jmjd3, which is essential for M2 microglia polarization, in the substantia nigra (SN) in vivo dramatically causes microglial overactivation and exacerbated dopamine (DA) neuron death in a PD animal model [35], indicating a protective role of M2 microglia in this process. However, long-term over-activation of microglia in the PD brain significantly up-regulates the expression of a large group of pro-inflammatory cytokines including TNF-α, IL-1β, interleukin-6 (IL-6) and IFN-γ, which contribute to the acceleration of nigral DA neuron degeneration [36, 37]. As the disease progresses, molecules such as α-synuclein, ATP and metalloproteinase-3 (MMP-3) released from the degenerating DA neurons will further enhance microglia activation, amplify the neuroinflammatory responses in the brain, and result in the deterioration of the neurodegenerative processes [11, 38] forming a vicious cycle of neurodegeneration. Activated microglia accumulate around the α-synuclein-positive aggregates in many regions of PD brain [39]. These cells are likely activated by over-produced [38, 40], mutants or misfolded α-synuclein leading to increases in the production and release of the pro-inflammatory cytokines [38, 41, 42]. Thus, the neurotoxicity induced by excessive or misfolded α-synuclein may be partially caused by microgliamediated inflammatory responses.
ATP, a purinergic neurotransmitter, is also able to robustly modulate various functions of microglia [43, 44]. The migration of microglia to the injured and inflammatory areas is controlled by ATP released from the damaged neurons and neighboring astrocytes [44]. In addition, ATP binds to the P2Y receptor which is mainly expressed by microglia in the brain and induces the production of high levels of IL-1β, TNF-α and nitric oxide (NO) [45]. Another protein produced by degenerating neurons is MMP-3 which also plays important roles in the regulation of the activation states of microglia, at least in vitro. Overexpression of MMP-3 in microglia-neuron co-cultures induces prominent activation of microglia and increased the oxidative stress reaction. In contrast, MMP-3−/− mice administrated with N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) display attenuated nigrostriatal DA neuronal degeneration, microglial activation, and superoxide generation [46]. These data support the notion that microglia is a major player in neuroinflammation in the context of PD pathogenesis and MMP-3 plays a pivotal role in dopaminergic neuronal degeneration.
Microglia activation phenotypes in PD
Mounting evidence indicates that microglia has two alternative activation phenotypes, termed the M1 (proinflammatory) phenotype and the alternative M2 (antiinflammatory) phenotype. These different activation statuses of microglia are characterized by secretion of different arrays of cytokines [47]. It has been demonstrated that LPS/IFN-γ treatment induces M1 activation, while IL-4/IL-13 treatment triggers M2 activation in microglia. The classical M1 activation of microglia is featured by the production of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-12, and other cytotoxic molecules such as superoxide, NO and reactive oxygen species (ROS), contributing to the amplification of the pro-inflammatory responses during injuries and infections. Conversely, M2 microglia plays an immunosuppressive role by antagonizing the classic M1 microglia and promoting tissue repair. The M2 microglia produces a variety of cytokines with antiinflammatory property, such as IL-4, IL-13, IL-10, and TGF-β. The different activation forms of microglia can be distinguished by their characteristic gene expression pattern. For example, Arg1, FIZZ1 (also known as RELM-α), Chi3l3 (also known as YM1) and CD206 were expressed in mouse M2 phase microglia [48]. Expression of Arg1, FIZZ1 and Chi3l3 may be regulated by cytokines, since their levels are significantly increased in primary cultured microglia or the striatal and frontal cortical regions of mouse brain following IL-4 stimulation [49].
What factors affect M1/M2 microglia phenotype in the context of PD? Mis-folded proteins and environmental toxins induce the activation of microglia toward M1 phenotype in PD animal models [38, 50, 51]. Chronic MPTP administration leads to progressive reduction of CD206 expression, which suggests the down-regulation of M2 phase activation of microglia in the progression of PD [47]. Conversely, IL-4 treatment up-regulates microglial expression of histone H3K27me3 demethylase (Jmjd3) which is involved in the regulation of epigenetic modification of chromosomes and contributes to various human diseases. Expression levels of M2 marker genes, such as Arg1 and CD206, are significantly down-regulated after Jmjd3 knockdown in N9 microglia cell line, indicating the essential role of Jmjd3 for M2 microglia polarization. Knockdown of Jmjd3 in vivo exacerbates the DA neuron loss in the SNpc of MPTP-induced mouse PD model by revoking M2 activation of microglia [35].
Conditioned medium (CM) from M1 phase N9 microglia results in increased death of DA neurons, whereas CM mixture from both M1 and M2 cells reverses the neurotoxicity elicited by the M1-CM [35]. Previous investigations indicated that a majority of the activated microglia express M2 associated genes at the early stages following injury in various models. M1 signature genes, however, gradually become predominant in later stages [32]. These interesting observations suggest that it is important to balance different microglia activation phenotypes in PD (Fig. 1). It appears to be a promising strategy to intervene the progression of PD by manipulating the transition of microglia activation statuses. Even though current data suggest differential role of M1 and M2 in the pathogenesis of PD in its animal models, similar results from patients are lacking. Future work should warrant research in this aspect.
چکیده
مقدمه
فنونیپ های فعال شدن میکروگلیا در بیماری پارکینسون
التهاب نورونی بواسطه ی استروسیت در بیماری پارکینسون
ژن های مربوط به بیماری پارکینسون و التهاب نورونی
التهاب ناشی از سلولهای ایمنی محیطی در بیماری پارکینسون
منابع
Abstract
Introduction
Microglia activation phenotypes in PD
Astrocyte-mediated neuroinflammation in PD
PD-associated genes and neuroinflammation
Peripheral immune cell-mediated inflammation in PD
References