
دانلود رایگان مقاله تجزیه و تحلیل ارزش پیش آگهی سرطان تخمدان
چکیده
سرطان تخمدان اولین دلیل ایجاد سرطان های مرگ آور زنان و زایمان است. برای شناسایی ژن های کلیدی و microRNA ها در سرطان تخمدان، مجموعه اطلاعات میکرواری mRNA، GSE36668,، GSE18520، GSE14407 و مجموعه اطلاعات microRNA GSE47841 از بیان ژن مجموعه اطلاعات Omnibus دانلود شده است. ژن های بیان شده DEG2 و میکروRna های (DEMs) با استفاده از GEO2R حاصل شدند. آنالیزهای عملکردی و غنی سازی مسیر برای DEGs با استفاده ازاطلاعات DAVID انجام شده است. برهمکنش شبکه پروتئین-پروتئین (PPI) توسط STRING انجام گرفت و توسط سیوسکوپ قابل دیدن شد. آنالیز بقای کلی (OS) ژن های قطبی با ابزار آنلاین Kaplan–Meier انجام شد. انالیز واحد شبکه PPI با استفاده از MCODE انجام گرفت. علاوه برآن miRecord برای پیشگویی هدف DEMS اجرا شد. نهایتا تعداد 345 DEMS تامین شد عمدتا با فرآیند سیکل سلولی، میتوز و تخمک گذاری در ارتباطند. یک شبکه PPI ، متشکل از 141 گره¬ و 269 edge ساخته شد. 16 تا از ژن ها درجه های بالایی در شبکه دارند. بیان بالا چهار ژن از 16 ژن با OS بیماران مبتلا به سرطان تخمدان، شامل CCNB1، CENPF، KIF11 و ZWINT مرتبط است. واحد قابل توجه از شبکه PPI کشف شده است. عملکردها و مسیرهای غنی شده شامل سیکل سلولی، تقسیم هسته و میتوز اووسیت است. به علاوه، مجموع 36 DEMs شناسایی شدند. بیان KIF11 به طور منفی با miR-424 و has-miR-381 ارتباط دارد و این همچنین یک هدف بالقوه microRNA ها است. در نتیجه، این ژن های کلیدی که میتوانند هدف های بالقوه برای تشخیص و درمان سرطان تخمدان را فراهم کنند، یافت میشود.
مقدمه
سرطان تخمدان کشنده ترین بیماری زنان و زایمان جهانی است با تخمین 238700 مورد جدید سرطان تخمدان و 151190 مرگ از این بیماری در هر سال. این مقدار بالای کشندگی سرطان تخمدان، عمدتا به علت نقصان روش¬های موثر تشخیصی در مراحل اولیه موش صحرایی است. با وجود ابهام علائم، 70% از بیماران، با سرطان تخمدان در مراحل پیشرفته تشخیص داده شده اند که نرخ بقای 5-ساله کمتر از 25 % در مقایسه با 90% بیماران با تومورهای تشخیص داده شده در فاز اولیه، دارند.
ماکرهای زیستی برای کاربرد تشخیص در سرطان تخمدان گسترش داده میشوند. به عنوان مثال، آنتی ژن 125 کربوهیدرات (CA-125) پرکاربردترین مارکر زیستی سرم برای سرطان تخمدان است. اما CA-125 در شرایط خوش خیم و بدخیم به شدت بیان میشود و تنها توسط 50% از سرطان های تخمدان مرحله ی اولیه مرتفع میشود. پروتئین 4 اپیدمی انسان (HE4) مارکرزیستی دیگری برای سرطان است که ویژگی بهتری دارد و نسبت به CA-125 کمتر حساس است. با این وجود، تاکنون، هیچ تکنولوژی شناسایی مناسبی با توجه به نقص تخصص یافتگی و حساسیت وجود ندارد. بر اساس این، ضروری است که مکانیسم های مولکولی در پیشرفت سرطان تخمدان بررسی شود و مارکرهای زیستی بیشتری برای تشخیص های موثرتر کشف شوند.
در سال های اخیر، تکنولوژی میکرواری به گستردگی برای تعیین تغییرات ژنتیکی عمومی در طول تولید تومورها استفاده شد. روش های بیوانفورماتیکی برای بررسی کردن مقدار زیادی از اطلاعات که توسط میکرواری حاصل میشود، ضروری است. در این کار، مثبت دروغین در نتیجه ی میکرواری ، سه مجموعه اطلاعات میکرواری mRNA و یک مجموعه اطلاعات microRNA برای حاصل شدن بیان ژن ها و microRNA ها بین بافت های سرطان تخمدان و نمونه های معمولی بافت انالیز شدند. آنالیز های شبکه ای و عملکردی برای شناسایی DEGs اجرا شدند که با آنالیزهای بقا و آنالیز برهمکنش mRNA-microRNA برای شناسایی ژن های کلیدی در سرطان تخمدان ترکیب شدند.
مواد و روش ها
اطلاعات میکرواری
Microarray بیان ژن Omnibus (GEO) یک مخزن عمومی برای ذخیره ی اطلاعات از قبیل میکرواری و توالی یابی نسل جدید است که بهصورت رایگان در دسترس کاربران است. پروفایل های بیان سه ژن (GSE36668, GSE18520 ,GSE14407 پروفایل بیان ژن miRNA GSE47841 از مجموعه اطلاعات GEO تامین شده است. اطلاعات میکرواری GSE36668 شامل نمونه های چهار بافت سرطان تخمدان و چهار نمونه های نرمال هستند. GSE18520 شامل 53 نمونه سرطان تخمدان و 10 نمونه نرمال هستند. GSE14407 شامل 12 نمونه ی سرطان تخمدان و 12 نمونه ی نرمال هستند . پروفایل بیان miRNA GSE47841 شامل 12 نمونه ی سرطان تخمدان و 9 نمونه ی نرمال است.
فرآوری داده ها
آرشیو مجموعه اطلاعات GEO ، تعداد بزرگی از مطالعات ژنومی عملکردی مقیاس بالا است که اطلاعاتی که توسط روش های گوناگون فرآوری و نرمال شده اند را شامل میشود. GEO2R برای micRNA ها و ژنهای بیان شده به طور مختلف در سرطان تخمدان و نمونه های نرمال اجرا شده است. GEO2R یک ابزار اینترنتی برهمکنشگر است که دو گروه از نمونه ها را تحت شرایط یکسان آزمایشگاهی با هم مقایسه میکند ومیتواند هر سری GEO را آنالیز کند. مقدار P تنظیم شده برای اصلاح نتایج مثبت دروغین با استفاده از روش نرخ یافتن Benjamini و Hochberg اجرا شده است. مقدار p<0.01 و |logFC|[1 تنظیم شده اند.
آنالیز عملکردی و غنای مسیر
مجموعه اطلاعات برای شرح نویسی ژن، قابل دیدار سازی و کشف قطعات ژن ورودی ، DAVID برنامه ای آنلاین است که مجموعه ی جامع از ابزار شرح نویسی ژن عملکردی برای محققان فراهم میکند تا مفهوم زیستی پشت مقادیر زیاد ژن ها را درک کنند. هستی شناسی ژن GO و ژن های Kyoto Encyclopedia و آنالیز غنی سازی مسیر ژنوم KEGG برای DEGs های شناخته شده با استفاده از مجموعه اطلاعات DAVID اجرا شد. P<0.05 تنظیم شده است.
ساخت شبکه PPI و انتخاب واحدها
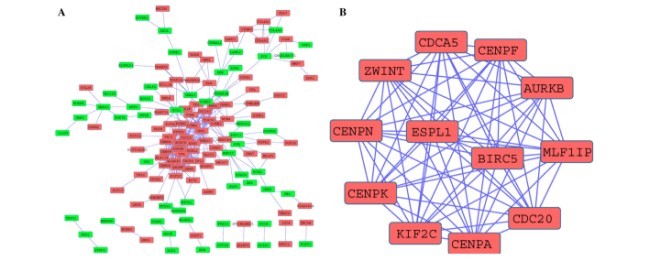
برهمکنش های عملکردی بین پروتئین ها میتواند زمینه ی مکانیسم مولکولی فرآیند سلولی را فراهم کند. در مطالعه ی اخیر شبکه ی برهمکنش پروتئین- پروتئین DEGs با استفاده از ابزار جستجوی برای بازیابی مجموعه اطلاعات ژن های کنشگر ساخته شده است. STRING و متعاقبا با استفاده از سیتوسکوپ قابل دیدار شده است. امتیاز اطمینان بزرگتر یا مساوی 0.7 تنظیم شده است. سپس کاوش مولکولی پیچیده MCODE برای واحدهای شبکه PPI با درجه انفصال =2، امتیاز گره= 0.2، هستهk=2، و ماکزیمم عمق=100 اجرا شده است. آنالیز غنای عملکردی ژن در هر واحد توسط DAVID انجام شده است.
پیشگویی هدف های micRNA
ژن های هدف micRNA بیان شده با استفاده از miRecords پیشگویی شده است، که یک منبع یکپارچه هستند که توسط 11 برنامه پیشگویی هدف micRNA تولید شده است. (DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, TargetScan) ژن هایی که توسط حداقل چها ربرنامه پیشگویی شده اند به عنوان هدف micRNA شناسایی شده اند.
Abstract
Ovarian cancer is the first leading cause of mortality in gynecological malignancies. To identify key genes and microRNAs in ovarian cancer, mRNA microarray dataset GSE36668, GSE18520, GSE14407 and microRNA dataset GSE47841 were downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) and microRNAs (DEMs) were obtained using GEO2R. Functional and pathway enrichment analysis were performed for DEGs using DAVID database. Protein–protein interaction (PPI) network was established by STRING and visualized by Cytoscape. Following, overall survival (OS) analysis of hub genes was performed by the Kaplan– Meier plotter online tool. Module analysis of the PPI network was performed using MCODE. Moreover, miRecords was applied to predict the targets of the DEMs. A total of 345 DEGs were obtained, which were mainly enriched in the terms related to cell cycle, mitosis, and ovulation cycle process. A PPI network was constructed, consisting of 141 nodes and 296 edges. Sixteen genes had high degrees in the network. High expression of four genes of the 16 genes was associated with worse OS of patients with ovarian cancer, including CCNB1, CENPF, KIF11, and ZWINT. A significant module was detected from the PPI network. The enriched functions and pathways included cell cycle, nuclear division, and oocyte meiosis. Additionally, a total of 36 DEMs were identified. The expression of KIF11 was negatively correlated with that of has-miR-424 and has-miR-381, and it was also the potential target of two microRNAs. In conclusion, these results identified key genes, which could provide potential targets for ovarian cancer diagnosis and treatment.
Introduction
Ovarian cancer is the most lethal gynecological malignancy worldwide, with an estimated 238,700 new cases of ovarian cancer and estimated 151,190 deaths from this disease every year [1]. The high mortality of ovarian cancer is mainly due to the lack of effective diagnostic methods at early stage and high recurrence rate [2]. With the vagueness of symptoms, approximately 70 % of patients are diagnosed with ovarian cancer at an advanced stage, in which the 5-year survival rate is less than 25 % compared to up to 90 % for patients diagnosed with this tumor in early prophase [3, 4].
Currently, molecular biomarkers have been developed for diagnostic use in ovarian cancer. For example, carbohydrate antigen-125 (CA-125) is the most extensively applied serum biomarker for ovarian cancer [5]. But CA125 is highly expressed in common benign and malignant conditions, and only elevated by about 50 % of early stage ovarian cancers [6, 7]. Human epididymis protein 4 (HE4) is another biomarker for ovarian cancer that has a greater specificity and a less sensitivity than CA-125 [8]. Nevertheless, to date, no detecting technology is suited to generable people due to the deficiency of specificity and sensitivity. Accordingly, it is crucial to investigate the molecular mechanisms in ovarian cancer progression and discover additional biomarkers for early effective diagnosis.
In recent years, the microarray technology has been broadly used to obtain general genetic alteration during tumorigenesis [9–12]. The bioinformatics methods are necessary to process a great deal of data generated by microarray technology. In this work, given the false positive in results of microarray, three mRNA microarray dataset and a microRNA dataset were analyzed to obtain differentially expressed genes (DEGs) and microRNAs (DEMs) between ovarian cancer tissues and normal tissues samples. Functional enrichment and network analysis were applied for identifying of DEGs, combined with survival analysis and mRNA–microRNA interaction analysis, to identify key genes in ovarian cancer.
Materials and methods
Microarray data
The Gene Expression Omnibus (GEO, http://www.ncbi. nlm.nih.gov/geo) is a public repository for data storage, such as microarray and next-generation sequencing, which is freely available to users. Three gene expression profiles (GSE36668, GSE18520 and GSE14407) and the miRNA expression profile of GSE47841 were obtained from GEO database. The array data of GSE36668 included four ovarian cancer tissue samples and four normal samples [10]. GSE18520 consisted of 53 ovarian cancer samples and ten normal samples [11]. GSE14407 included 12 ovarian cancer samples and 12 normal samples [9]. The miRNA expression profile of GSE47841 included 12 ovarian cancer samples and nine normal samples [12].
Data processing
The GEO database archives a large number of highthroughput functional genomic studies that contain data that are processed and normalized using various methods. GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) was applied to screen differentially expressed miRNAs and genes between ovarian cancer and normal samples. GEO2R is an interactive web tool that compares two groups of samples under the same experimental conditions and can analyze almost any GEO series [13]. The adjusted P values (adj. P) were applied to correct for the occurrence of false positive results using Benjamini and Hochberg false discovery rate method by default. The adj. P\ 0.01 and |logFC| [ 1 were set as the cut-off criterion.
Functional and pathway enrichment analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) is an online program that provides a comprehensive set of functional annotation tools for researchers to understand biological meaning behind plenty of genes [14]. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed for identified DEGs using DAVID database. P\ 0.05 was set as the cut-off criterion.
PPI network construction and modules selection
The functional interactions between proteins can provide context in molecular mechanism of cellular processing. In present study, protein–protein interaction (PPI) network of DEGs was constructed using the Search Tool for the Retrieval of Interacting Genes (STRING, http://string. embl.de/) database and subsequently was visualized using Cytoscape [15, 16]. And confidence score C0.7 was set as the cut-off criterion. Then, the Molecular Complex Detection (MCODE) was performed to screen modules of PPI network with degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and max. depth = 100 [17]. The functional enrichment analysis of genes in each module was performed by DAVID.
Prediction of miRNA targets
The target genes of differentially expressed miRNAs were predicted using miRecords (http://c1.accurascience.com/ miRecords/), which is an integrated resource produced by 11 established miRNA target prediction programs (DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, and TargetScan) [18]. The genes predicted by at least four programs were identified as the targets of miRNAs.
چکیده
مقدمه
مواد و روش ها
فرآوری داده ها
پردازش داده ها
آنالیز عملکردی و غنای مسیر
ساخت شبکه PPI و انتخاب واحدها
پیشگویی هدف های micRNA
آنالیز بقای DEGs
نتایج
شناسایی DEGs
آنالیز عملکردی و غنای مسیر
ساخت شبکه PPI و انتخاب واحدها
جفت های miRNA-DEG
Kaplan–Meier plotter
بحث
منابع
Abstract
Introduction
Materials and methods
Microarray data
Data processing
Functional and pathway enrichment analysis
PPI network construction and modules selection
Prediction of miRNA targets
Survival analysis of DEGs
Results
Identification of DEGs
Functional and pathway enrichment analysis
PPI network construction and modules selection
miRNA–DEG
The Kaplan–Meier plotter
Discussion
References